
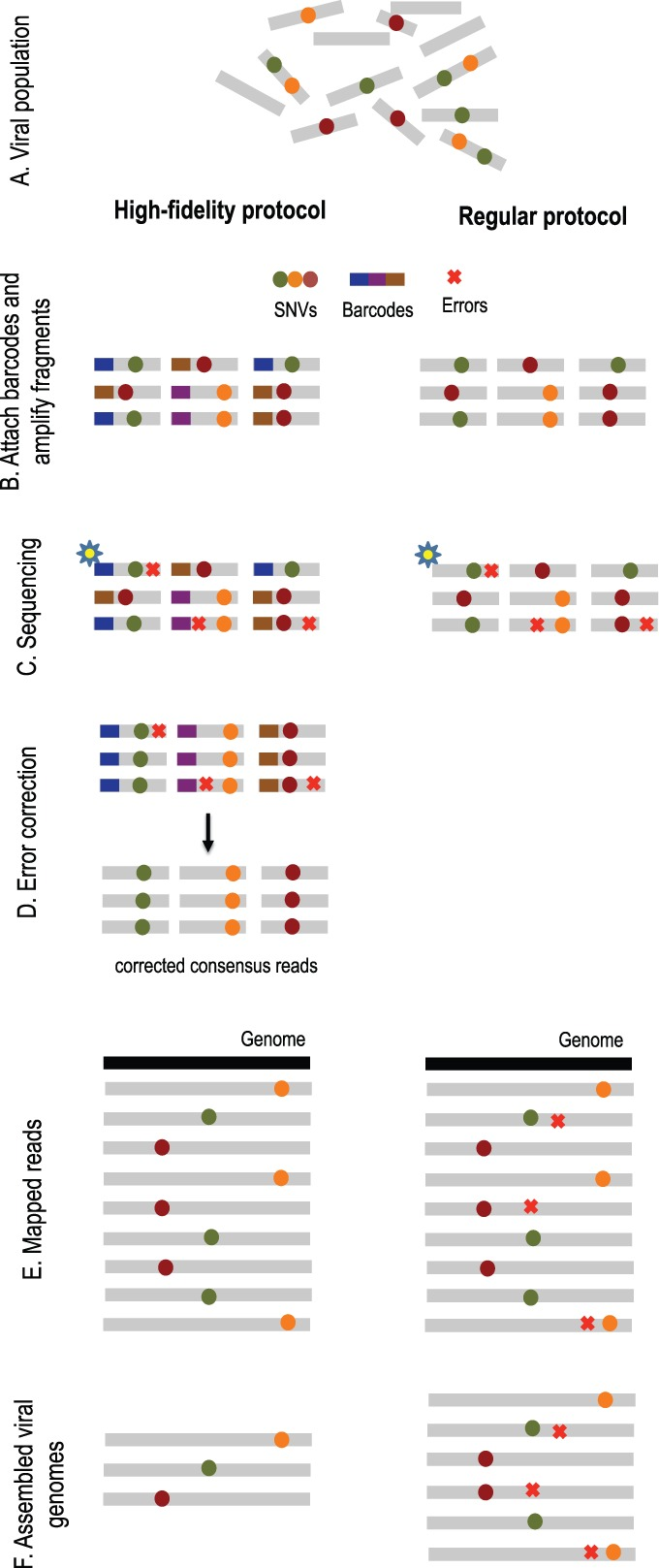
Overview of high-fidelity sequencing protocol. (A) DNA material from a viral population is cleaved into sequence fragments using any suitable restriction enzyme. (B) Individual barcode sequences are attached to the fragments. Each tagged fragment is amplified by the polymerase chain reaction (PCR). (C) Amplified fragments are then sequenced. (D) Reads are grouped according to the fragment of origin based on their individual barcode sequence. An error-correction protocol is applied for every read group, correcting the sequencing errors inside the group and producing corrected consensus reads. (E) Error-corrected reads are mapped to the population consensus. (F) SNVs are detected and assembled into individual viral genomes. The ordinary protocol lacks steps (B) and (D)
Viral populations change rapidly throughout the course of an infection. Due to this, drugs that initially control an infection can rapidly become ineffective as the viral drug target mutates. For better drug design, however, we first must develop techniques to be able to detect and quantify the presence of various rare viral variants from a given sample.
Currently, next-generation sequencing technologies are employed to better understand and quantify viral population diversity. The existing technologies, however, have difficulty if distinguishing between rare viral variants and sequencing errors even when sequencing with high coverage. To overcome this problem, our lab has proposed a two-step solution in a recent paper by Serghei Mangul.
In his paper, Serghei suggested we first use a high-fidelity protocol known as Safe-SeqS with high coverage. This method employs the use of small individual barcodes that are attached to sequencing fragments before undergoing amplification by polymerase chain reaction (PCR) and being sequenced. By comparing and taking a consensus of amplicons from the same initial sequence fragment, we can easily eliminate some sequencing errors from our data.
These consensus reads then are assembled using an accurate viral assembly method Serghei developed known as the Viral Genome Assembler (VGA). This software uses read overlapping, SNV detection, and a conflict graph to distinguish and reconstruct genome variants in the population. Finally, an expectation-maximization algorithm is used to estimate abundances of assembled viral variants.
In the paper, this approach was applied to both simulated and real data and found to outperform current state-of-the-art methods. Additionally, this viral assembly method is the first of its kind to scale to millions of sequencing reads.
The Viral Genome Assembler tool is freely available here: http://genetics.cs.ucla.edu/vga/
From the paper:
Advances in NGS and the ability to generate deep coverage data in the form of millions of reads provide exceptional resolution for studying the underlying genetic diversity of complex viral populations. However, errors produced by most sequencing protocols complicate distinguishing between true biological mutations and technical artifacts that confound detection of rare mutations and rare individual genome variants. A common approach is to use post-sequencing error correction techniques able to partially correct the sequencing errors. In contrast to clonal samples, the post-sequencing error correction methods are not well suited for mixed viral samples and may lead to filtering out true biological mutations. For this reason, current viral assembly methods are able to detect only highly abundant SNV, thus limiting the discovery of rare viral genomes.Additional difficulty arises from the genomic architectures of viruses. Long common regions shared across viral population (known as conserved regions) introduce ambiguity in the assembly process. Conserved regions may be due low-diversity population or due to recombination with multiple cross-overs. In contrast to repeats in genome assembly, conserved regions may be phased based on relative abundances of viral variants. Low-diversity viral populations in which all pairs of individual genomes within a viral population have a small genetic distance from each other may represent additional challenges for the assembly procedure.
We apply a high-fidelity sequencing protocol to study viral population structure (Fig. 1). This protocol is able to eliminate errors from sequencing data by attaching individual barcodes during the library preparation step. After the fragments are sequenced, the barcodes identify clusters of reads that originated from the same fragment, thus facilitating error correction. Given that many reads are required to sequence each fragment, we are trading off an increase in sequence coverage for a reduction in error rate. Prior to assembly, we utilize the de novo consensus reconstruction tool, Vicuna (Yang et al., 2012), to produce a linear consensus directly from the sequence data. This approach offers more flexibility for samples that do not have ‘close’ reference sequences available. Traditional assembly methods (Gnerre et al., 2011; Luo et al., 2012; Zerbino and Birney, 2008) aim to reconstruct a linear consensus sequence and are not well-suited for assembling a large number of highly similar but distinct viral genomes. We instead take our ideas from haplotype assembly methods (Bansal and Bafna, 2008; Yang et al., 2013), which aim to reconstruct two closely related haplotypes. However, these methods are not applicable for assembly of a large (a priori unknown) number of individual genomes. Many existing viral assemblers estimate local population diversity and are not well suited for assembling full-length quasi-species variants spanning the entire viral genome. Available genome-wide assemblers able to reconstruct full-length quasi-species variants are originally designed for low throughput and are impractical for high throughput technologies containing millions of sequencing reads.
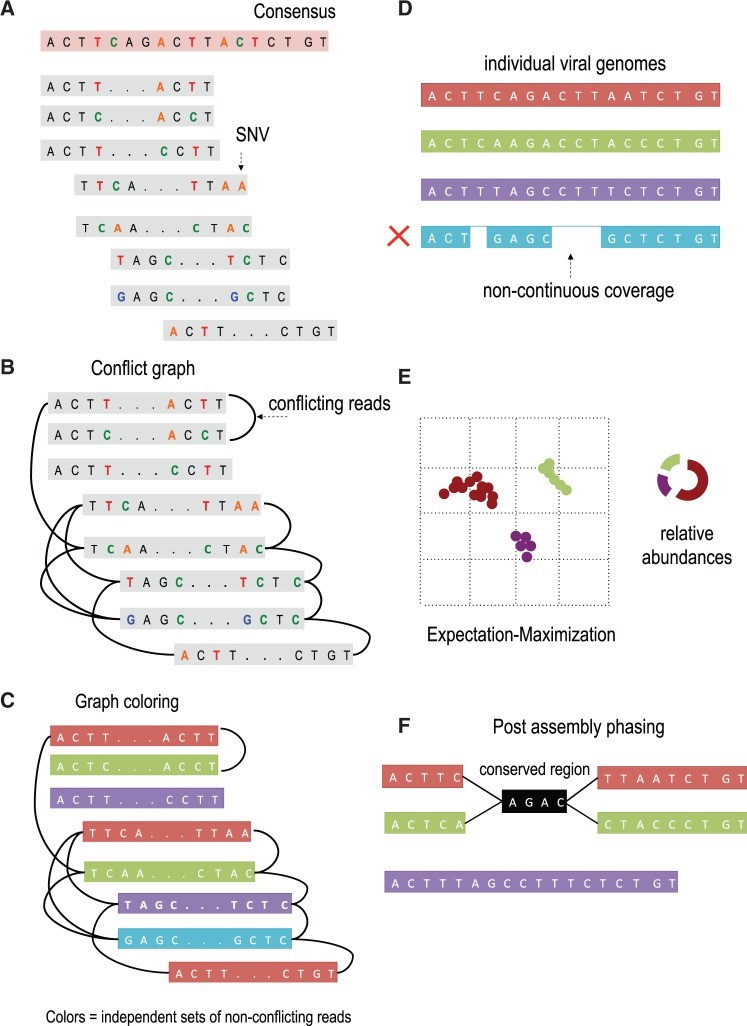
Overview of VGA. (A) The algorithm takes as input paired-end reads that have been mapped to the population consensus. (B) The first step in the assembly is to determine pairs of conflicting reads that share different SNVs in the overlapping region. Pairs of conflicting reads are connected in the ‘conflict graph’. Each read has a node in the graph, and an edge is placed between each pair of conflicting reads. (C) The graph is colored into a minimal set of colors to distinguish between genome variants in the population. Colors of the graph correspond to independent sets of non-conflicting reads that are assembled into genome variants. In this example, the conflict graph can be minimally colored with four colors (red, green, violet and turquoise), each representing individual viral genomes. (D) Reads of the same color are then assembled into individual viral genomes. Only fully covered viral genomes are reported. (E) Reads are assigned to assembled viral genomes. Read may be shared across two or more viral genomes. VGA infers relative abundances of viral genomes using the expectation–maximization algorithm. (F) Long conserved regions are detected and phased based on expression profiles. In this example red and green viral genome share a long conserved region (colored in black). There is no direct evidence how the viral sub-genomes across the conserved region should be connected. In this example four possible phasing are valid. VGA use the expression information of every sub-genome to resolve ambiguous phasing.
We introduce a viral population assembly method (Fig. 2) working on highly accurate sequencing data able to detect rare variants and tolerate conserved regions shared across the population. Our method is coupled with post-assembly procedures able to detect and resolve ambiguity raised from long conserved regions using expression profiles (Fig. 2F). After a consensus has been reconstructed directly from the sequence data, our method detects SNVs from the aligned sequencing reads. Read overlapping is used to link individual SNVs and distinguish between genome variants in the population. The viral population is condensed in a conflict graph built from aligned sequencing data. Two reads are originated from different viral genomes if they share different SNVs in the overlapping region. Viral variants are identified from the graph as independent sets of non-conflicting reads. Non-continuous coverage of rare viral variants may limit assembly capacities, indicating that increase in coverage is required to increase the assembly accuracy. Frequencies of identified variants are then estimated using an expectation–maximization algorithm. Compared with existing approaches, we are able to detect rare population variants while achieving high assembly accuracy.
The full citation of our paper is:
