
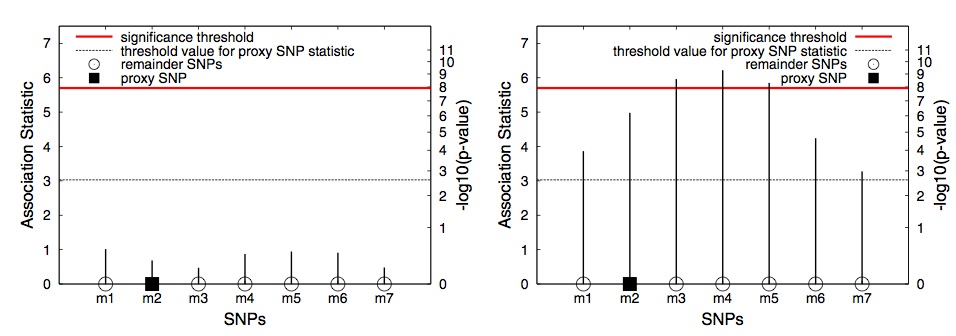
Fig. 1. An example of applying GRAT in two hypothetical regions. First, the proxy SNP (rectangle) is tested and its statistics is compared to the threshold (dashed line). If the statistic is above the threshold, the remaining SNPs in the region are tested.
Emrah Kostem in our group has recently published a new method for association analysis targeted toward eQTL analysis called GRAT(10.1007/978-3-642-37195-0_10). GRAT is designed to speed up eQTL studies and the software is available at http://genetics.cs.ucla.edu/GRAT
Over the past few years, the genome wide association study (GWAS) approach has been applied to identify regions of the genome, which harbor genetic variation that affects gene expression levels. These regions are referred to as expression quantitative trait loci (eQTL)(10.1038/nrg2537).(10.1038/nrg1964). In a typical eQTL study, the GWAS approach is applied to tens of thousands of gene expression levels using millions of SNPs, resulting in billions of association statistics to be computed. This results in a tremendous computational burden, which is only increasing with sequencing technology collecting more genetic variations and high-throughput genomic data collecting more phenotypic data such as isoform expression(10.1016/j.tig.2010.10.006). This problem is compounded by the fact that some of the statistical techniques for analyzing eQTLs utilize mixed models and themselves are computationally expensive(10.1038/ng.548),(10.1038/nmeth.1681).(10.1038/ng.2310).
We recently published a paper on a method, GRAT, to perform association analysis in high-throughput phenotype datasets, such as the eQTL studies.
The key idea behind GRAT is that we first test a subset of the SNPs and only in regions where the statistic is above a threshold, we test the remaining regions. In contrast to testing all SNPs, our approach tests around 10% of the SNPs in two-stages and guarantees to identify all significant associations with a very high accuracy.
Here is a description of our method from the paper:
Genome-Wide Rapid Association Testing (GRAT)
In Figure 1, we consider two possible scenarios for a genomic region in a GWAS. In (a) the region contains no significant associations and in (b) the region con- tains a causal SNP. In (a) and (b), the statistics for each SNP are shown, denoting what could have been observed in each scenario had all the SNPs in the region been tested. Let m2 be the proxy SNP for this region to decide whether or not to test the rest of the SNPs. We refer to the SNPs other than the proxy SNP ( m1, m3, m4, m5, m6 and m7 ) as the “remainder SNPs”. If the observed statistic of the proxy SNP is stronger than a threshold value, which in this example is 3.0, the remainder SNPs are tested.
In the first-stage, only the proxy SNP is tested and its association statistic is observed. In (a), where the region contains no associations, the statistic of the proxy SNP is 0.7. The observed statistic of the proxy is less than the threshold value ( 0.7 < 3.0 ) and hence none of the remainder SNPs within the region are tested. In (b), the region contains associations and the proxy SNP captures this information. The observed statistic of the proxy SNP is stronger than the thresh- old value ( 5.0 > 3.0 ), which leads to testing each of the remainder SNPs in the region. This results in identifying all the significant SNPs ( m3, m4 and m5 ).
In the paper, we introduce a novel approach for choosing the proxy SNPs and the threshold values, which provide guarantees that all statistically significant associations will be discovered while computing the least amount of association tests. Due to the complexity of linkage disequilibrium (LD) across the genome, we use a separate threshold value for each remainder SNP rather than using a common threshold value for all the remainders SNPs in an LD region. This is performed by pairing each remainder SNP with its most strongly correlated proxy SNP and a threshold value is used for the pair to decide whether or not to test the remainder SNP. We have precomputed the proxy SNPs for the 1000 Genomes Project and studies imputing to SNPs in this reference can benefit from our method. Even though the LD structure among the SNPs in the study and the reference dataset may be different, our method guarantees to discover all significant associations with high-probability. This is achieved by updating the threshold values using the LD structure observed in the study. We term our novel two-stage testing procedure as Genome-wide Rapid Association Testing (GRAT).
GRAT can be applied to a wide range of statistical models, such as case- control studies, quantitative traits and linear mixed models (LMM). In particu- lar, the LMM approach has recently become popular due to its effective control of population structure. Computing the LMM association statistic is compu- tationally expensive and recently its efficient computation has attracted great interest(10.1038/ng.548),(10.1038/nmeth.1681).(10.1038/ng.2310). The speed-up due to GRAT is cumulative with these efforts.
There are some interesting aspects to the computational method. For given proxy SNPs, our method solves an optimization problem that minimizes the number of SNPs tested that will result in a given recall rate. We prove that this problem is convex and show that it can be solved very efficiently.
Furthermore, we propose a greedy algorithm to search for the optimal proxy SNPs.
The full citation is:
