Our group, in an effort led by former UCLA PhD student Dan He, developed an algorithm for reconstructing pedigrees with genotype data. This novel approach is presented in a paper recently published in IEEE/ACM Transactions on Computational Biology and Bioinformatics.
Pedigree inference plays an important role in population genetics. Pedigrees, commonly known as family trees, represent genetic relationships between individuals of a family. A pedigree diagram provides a model to compute the inheritance probability for the observed genotype and encodes all possible inheritance options for an allele in an individual. Pedigree reconstruction methods face several challenges. First, there can be an exponential number of possible pedigree graphs, and, second, the number of unknown ancestors can become very large as the height of the pedigree increases.
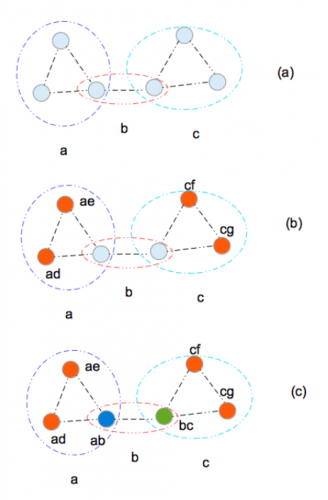
Examples of sequentially labeling the half-sibling graph. For more information, see our paper.
Our project uses genotype data to reconstruct pedigrees with computational efficiency despite these challenges. Our previous method, IPED, is the only known algorithm scalable to large pedigrees with reasonable accuracy for cases involving both outbreeding and inbreeding. IPED starts from extant individuals and reconstructs the pedigree generation by generation backwards in time. For each generation, IPED predicts the pairwise relationships between the individuals at the current generation and create parents for them according to their relationships.
Existing methods, including IPED, only consider pedigrees with simple structure; they cannot handle populations where, for example, two children share only one parent. To improve pedigree reconstruction when populations have complex structure, we proposed the novel method IPED2. Our approach uses a new statistical test to detect half-sibling relationships and a new graph-based algorithm to reconstruct the pedigree when half-siblings are allowed.
In order to test the performance of our method on complicated pedigrees, we use simulated pedigrees with different parameter settings and, instead of genotype data, we simulate haplotypes
directly. Our experiments show that IPED2 outperforms IPED and two other existing approaches for cases where there are half-siblings.
To our knowledge, this is the first method that can, using just genotype data, reconstruct pedigrees with half-siblings and inbreeding. IPED2 is also scalable to large pedigrees. In future work, we would like to consider additional genetic actions, such as insertion, deletion, and replacement, to resolve the conflicts. We also plan to refine IPED2 to consider cases where genotypes of ancestral individuals are known and where genotypes of extant individuals that are not on the lowest generations are known.
For more information, see our paper, which is available for download through Bioinformatics: http://ieeexplore.ieee.org/abstract/document/7888513/.
In addition, the open source implementation of IPED2, which was developed by Dan He, is freely available for download at http://genetics.cs.ucla.edu/Dan/Software/IPED2.html.
The full citation to our paper is:
He, D., Wang, Z., Parida, L. and Eskin, E., 2017. IPED2: Inheritance path based pedigree reconstruction algorithm for complicated pedigrees. IEEE/ACM Transactions on Computational Biology and Bioinformatics.