Microbial communities inhabiting the human body exhibit significant variability across different individuals and tissues and are likely play an important role in health and disease. Serghei Mangul and David Koslicki (Oregon State University) recently published a paper presenting a novel approach for characterizing microbial communities in metatranscriptomics studies. Koslicki developed this tool, which may help scientists explore the role microbiota play in disease development, especially when comparing microbiomes of healthy and disease subjects.
Identifying and characterizing the relative abundance of microbiota in different tissues is essential to better understanding the role of microbial communities in human health. Current approaches use reference databases to identify, classify, and compare microbial communities present in the individual host. However, existing databases are incomplete and rely on a limited compendium of reference genomes. Current reference-based approaches are unable to accurately determine microbial compositions to the extent that could be possible given the high resolution of data produced by today’s high throughput sequencing technology.
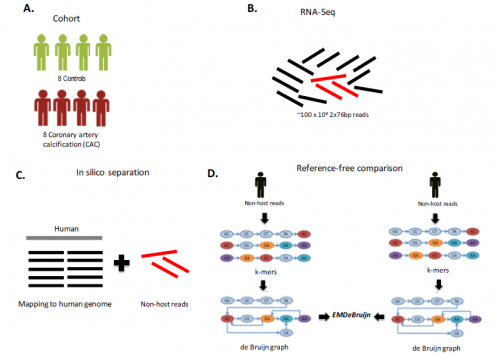
Framework of the study. For more information, download our paper.
Ideally, comparison of microbial communities across samples could circumvent this limiting classification step. Mangul and Koslicki recently developed EMDeBruijn, a reference-free approach that uses all available non-host microbial reads, not just those classified in reference databases, to compare microbial communities.
First, EMDeBruijn translates sequencing data to a de Bruijn graph, which represents overlaps between symbols in sequences. De Bruijn graphs are commonly used in de novo assembly of short read sequences to a genome, but have not yet been applied in a reference-free approach. EMDeBruijn then uses properties of the de Bruijn graphs to compare microbiome composition across individuals. This metric is reduced using the Earth Mover’s Distance (EMD), a statistic that can measure the distance between two probability distributions over a region.
In their recent paper, Mangul and Koslicki applied EMDeBruijn to study the composition and abundance levels of the microbial communities present in blood samples from coronary artery calcification (CAC) patients and controls. EMDeBruijn uses candidate microbial reads to differentiate between case (CAC-affected) and control (healthy) samples, and a filtered set of non-host reads are used to determine the composition of the blood microbiome. Hierarchical clustering using the EMDeBruijn metric successfully identifies several large clusters unique to samples from either health or control groups.
This study indicates the presence of the disease-specific microbial community structure in CAC patients, and points to the need for additional investigation of potentially causal relationships between the microbiome and CAC disease.
Using the same data set, Mangul and Koslicki compare the results of EMDeBruijn with those of current approaches. Existing computational methods, including MetaPhlAn and RDP’s NBC, discovered various microbial communities across the health and control samples. However, neither of these methods were able to identify any disease-specific patterns in the microbiome nor discriminate the samples into disease and healthy groups.
EMDeBruijn provides a powerful, species independent way to assess microbial diversity across individuals and subjects. For more information, see our paper, which was published in the Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics: http://dl.acm.org/citation.cfm?id=2975174.
Code implementing this method is available at: https://github.com/dkoslicki/EMDeBruijn.

Visualization of the EMDeBruijn Distance. a) Pictorial representation of 2-mer frequencies for two hypothetical samples, S1 and S2. b) The 2-mer frequencies overlaid the de Bruijn graph B2(A ). c) Representation of the flow used to compute EMD2(S1; S2); dark arrows denote mass moved from the initial node to the terminal node. d) Result of applying the flow to the 2-mer frequencies of S1.
This project was a collaboration that started at the Mathematical and Computational Approaches in High-Throughput Genomics program held in Fall 2011 at the Institute of Pure and Applied Mathematics (IPAM). Our on-going Computational Genomics Summer Institute (CGSI; also co-organized by IPAM) was inspired by the 2011 program. Check out the 2017 CGSI website for a preview of this summer’s programs – the deadline for applications is February 1, 2017!
The full citation to our paper is:
Mangul S, Koslicki D. Reference-free comparison of microbial communities via de Bruijn graphs. In Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. 2016 Oct 2 (pp. 68-77). Association for Computing Machinery, New York.