Bioinformatics is a rapidly growing field comprised of multiple academic disciplines. The work of quantitative geneticists is often not well understood by scholars conducting other types of research in Genetics. In response to this information gap, we are launching a series of reviews that are aimed to make common problems in computational biology research accessible to anyone in Genetics. We hope these reviews help researchers in Genetics better understand the scope and applicability of each other’s work, and serve as study guides for students taking college courses on the subject matter.
Today we made available on bioRxiv the first paper in this series, our review of population structure and relatedness in association studies. A genome-wide association study (GWAS) seeks to identify genetic variants that contribute to the development and progression of a specific disease. Over the past 10 years, new approaches using mixed models have emerged to mitigate the deleterious effects of population structure and relatedness in association studies. However, developing GWAS techniques to effectively test for association while correcting for population structure is a computational and statistical challenge. Our review motivates the problem of population structure in association studies using laboratory mouse strains and how it can cause false positives associations. We then motivate mixed models in the context of unmodeled factors.
To read the full review, download our paper: http://biorxiv.org/content/early/2016/12/07/092106.
This review was written by Lana Martin and Eleazar Eskin. We welcome feedback; please e-mail Lana if you have comments or questions: lana [dot] martin [at] ucla [dot] edu.
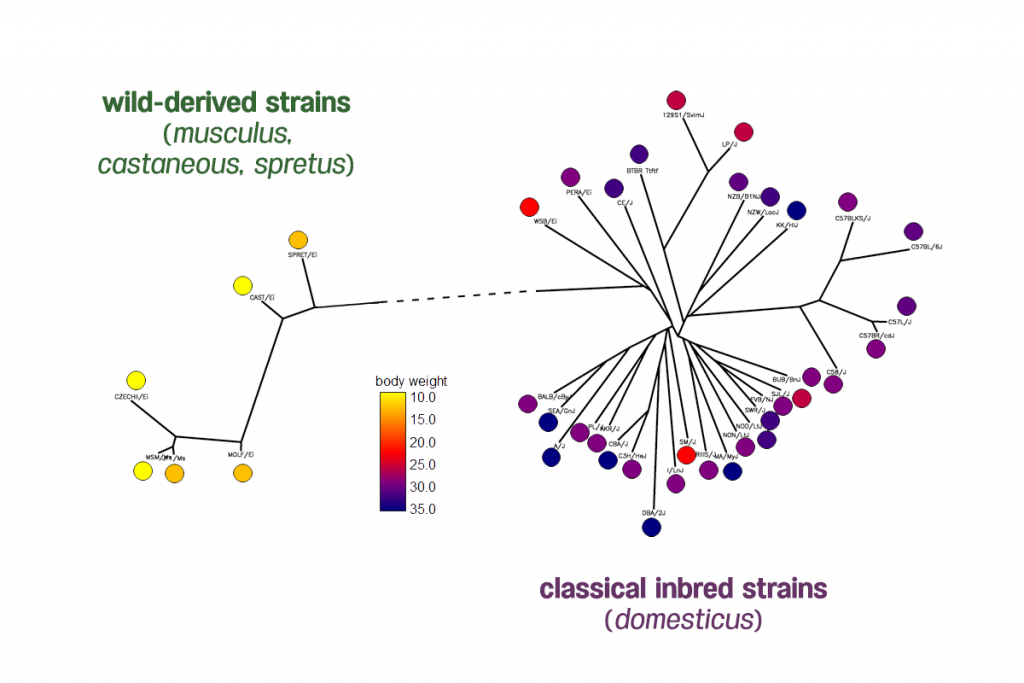
Body weight phenotypes of 38 inbred mouse strains from the Mouse Phenome Database generated by The Jackson Laboratory. The distribution of mice body weights shows two clades of mice have very different body weights.