
Recent advances in RNA sequencing technology can generate deep coverage data containing millions of reads. RNA-Seq data are used to identify genetic variants and alternatively spliced isoforms, a common mechanism for diversity in a gene, that may play a role in heritable traits and diseases. Using this type of data, connections can be drawn between genetic expression and one of the two parental haplotypes identified in a diploid organism’s transcript. In other words, we can potentially identify the parent from which an individual inherited a group of genes.
These multi-kilobase reads are longer than most transcripts and enable sequencing of complete haplotype isoforms. New computational methods are required for efficient analysis of this highly complex data. In a recent paper, we present HapIso (Haplotype-specific Isoform Reconstruction), a comprehensive method that can accurately reconstruct the haplotype-specific isoforms of a diploid cell. Our software package is the first method capable of reconstructing the haplotype-specific isoforms from long single-molecule reads.
HapIso uses splice mapping of long single-molecule reads to partition reads into two parental haplotypes. The single molecule reads entirely span the RNA transcripts and bridge the single nucleotide variation (SNV) loci across a single gene. To overcome gapped coverage and splicing structures of the gene, the haplotype reconstruction procedure is applied independently to regions of contiguous coverage that have been defined as transcribed segments. Restricted reads from the transcribed regions are partitioned into two local clusters using the 2-mean clustering. Using the linkage provided by the long single-molecule reads, we connect the local clusters into two global clusters. An error-correction protocol is then applied for the reads from the same cluster.
Discriminating the long reads into parental haplotypes allows HapIso to accurately calculate allele-specific gene expression and identify imprinted genes. Additionally, it has a potential to improve detection of the effect of cis– and trans-regulatory changes on gene expression regulation. Long reads allow access to genetic variation in regions previously unreachable by short read protocols and potentially lead to new insights in disease heritability.
We applied HapIso to publicly available single-molecule RNA-Seq data from the GM12878 cell line and circular-consensus (CCS) single-molecule reads generated by Pacific Biosciences platform. Our method discovered novel SNVs in regions that were previously unreachable by standard short read protocols, 53% of which follow Mendelian inheritance. HapIso detected 921 genes with both haplotypes expressed among 9,000 expressed genes. We observed 4,140 heterozygous loci corresponding to positions with non-identical alleles among inferred haplotypes. Additionally, we can theoretically identify recombinations in the transmitted haplotypes by checking the number of recombinations in the inferred haplotypes.
The open source Python implementation of HapIso was developed by Serghei Mangul and Harry (Taegyun) Yang, and the software package is freely available for download at https://github.com/smangul1/HapIso/.
This paper appears in Proceedings of the International Symposium on Bioinformatics Research and Applications (ISBRA-2016), which can be downloaded here: http://link.springer.com/chapter/10.1007%2F978-3-319-38782-6_7
Serghei Mangul and Harry Yang led this project, which involved Farhad Hormozdiari. The full citation to our paper is:
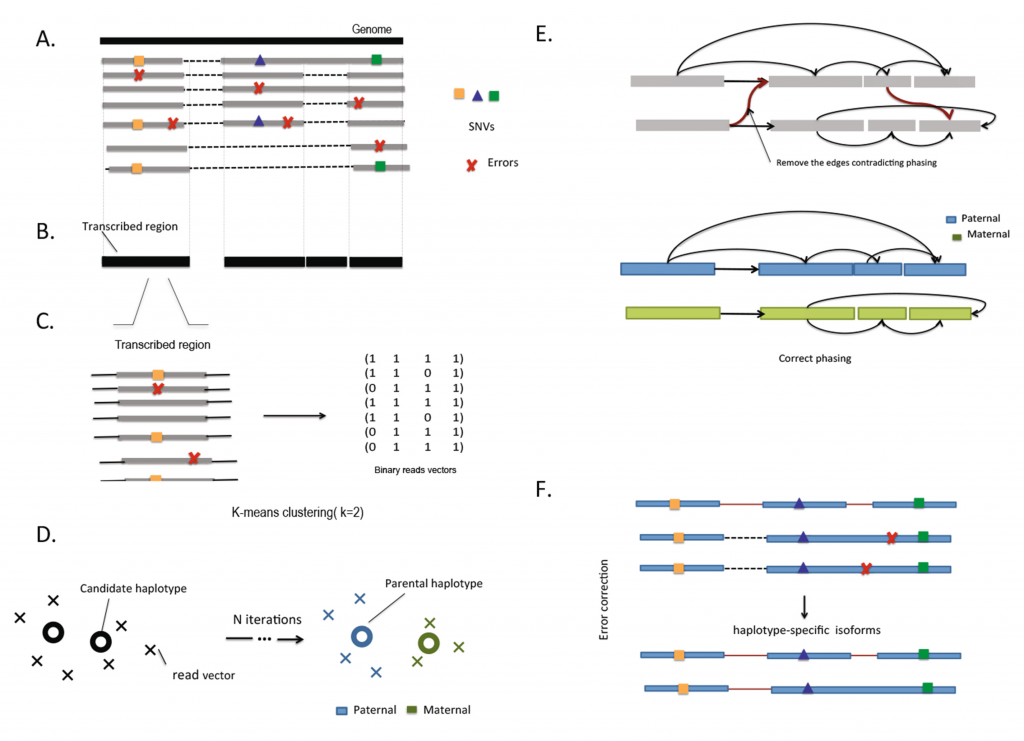
Overview of HapIso.
