
Although genome-wide association studies (GWAS) have identified thousands of variants associated with common diseases and complex traits, only a handful of these variants are validated to be causal. We consider ‘causal variants’ as variants which are responsible for the association signal at a locus. As opposed to association studies that benefit from linkage disequilibrium (LD), the main challenge in identifying causal variants at associated loci lies in distinguishing among the many closely correlated variants due to LD. This is particularly important for model organisms such as inbred mice, where LD extends much further than in human populations, resulting in large stretches of the genome with significantly associated variants. Furthermore, these model organisms are highly structured and require correction for population structure to remove potential spurious associations.
In our recently published work, we propose CAVIAR-Gene (CAusal Variants Identification in Associated Regions), a novel method that is able to operate across large LD regions of the genome while also correcting for population structure. A key feature of our approach is that it provides as output a minimally sized set of genes that captures the genes which harbor causal variants with probability q. Through extensive simulations, we demonstrate that our method not only speeds up computation, but also have an average of 10% higher recall rate compared with the existing approaches. We validate our method using a real mouse high-density lipoprotein data (HDL) and show that CAVIAR-Gene is able to identify Apoa2 (a gene known to harbor causal variants for HDL), while reducing the number of genes that need to be tested for functionality by a factor of 2.
In the context of association studies, the genetic variants which are responsible for the association signal at a locus are referred to in the genetics literature as the ‘causal variants.’ Causal variants have biological effect on the phenotype.
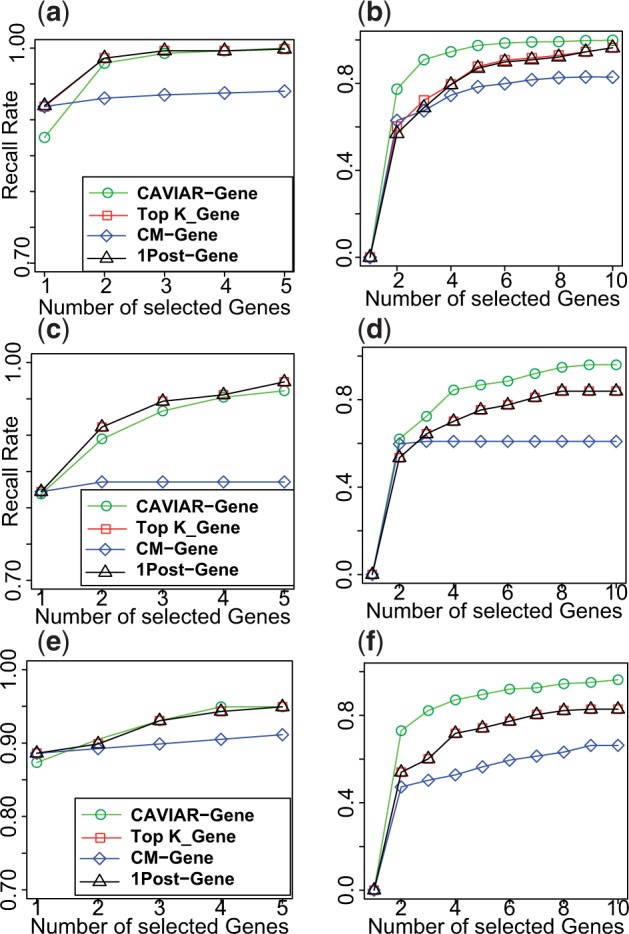
CAVIAR-Gene provides better ranking of the causal genes for Outbred, F2, and HMDP datasets. Panels a and b illustrate the results for Outbred genotypes for case where we have one causal and two causal genes, respectively. Panels c and d illustrate the results for F2 genotypes for case where we have one causal and two causal genes, respectively. Panels e and f illustrate the results for Outbred genotypes for case where we have one causal and two causal genes, respectively.
Generally, variants can be categorized into three main groups. The first group is the causal variants which have a biological effect on the phenotype and are responsible for the association signal. The second group is the variants which are statistically associated with the phenotype due to LD with a causal variant. Even though association tests for these variants may be statistically significant, under our definition, they are not causal variants. The third group is the variants which are not statistically associated with the phenotype and are not causal.
CAVIAR-Gene is a statistical method for fine mapping that addresses two main limitations of existing methods. First, as opposed to existing approaches that focus on individual variants, we propose to search only over the space of gene combinations that explain the statistical association signal, and thus drastically reduce runtime. Second, CAVIAR-Gene extends existing framework for fine mapping to account for population structure. The output of our approach is a minimal set of genes that will contain the true casual gene at a pre-specified significance level. The output of our approach is a minimal set of genes that will contain the true casual gene at a pre-specified significance level. This gene set together with its individual gene probability of causality provides a natural way of prioritizing genes for functional testing (e.g. knockout strategies) in model organisms. Through extensive simulations, we demonstrate that CAVIAR-Gene is superior to existing methodologies, requiring the smallest set of genes to follow-up in order to capture the true causal gene(s).
Building off our previous work with CAVIAR, CAVIAR-Gene takes as input the marginal statistics for each variant at a locus, an LD matrix consisting of pairwise Pearson correlations computed between the genotypes of a pair of genetic variants, a partitioning of the set of variants in a locus into genes, and the kinship matrix which indicates the genetic similarity between each pair of individuals. Marginal statistics are computed using methods that correct for population structure. We consider a variant to be causal when the variant is responsible for the association signal at a locus and aim to discriminate these variants from ones that are correlated due to LD.
In model organisms, the large stretches of LD regions result in a large number of variants associated in each region, thus making CAVIAR computationally
infeasible. Instead of producing a rho causal set of SNPs, CAVIAR-gene detects a ‘q causal gene set’ which is a set of genes in the locus that will contain the actual causal genes with probability of at least q.
For further details of our new method, CAVIAR-gene, view our full paper here:

I’m curious if this method is still unbiased if GWAS effects were estimated from mixed models.