
 Mixed models are now widely used for association studies in order to correct for population structure. A simple intuitive description of how and why they work is provided in our Mouse GWAS review(10.1038/nrg3335) paper published in Nature Genetics as a Box 1 on page 812:
Mixed models are now widely used for association studies in order to correct for population structure. A simple intuitive description of how and why they work is provided in our Mouse GWAS review(10.1038/nrg3335) paper published in Nature Genetics as a Box 1 on page 812:
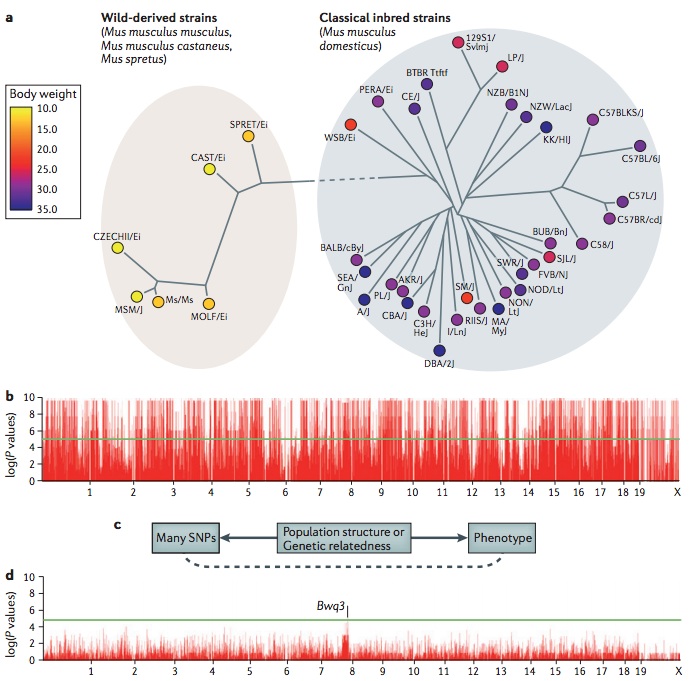
A challenge in mouse genome-wide association studies (GWASs) is the complex genetic relationships between strains included in the study. Some of these differences stem from the distinct ancestral origins of the mice, such as the differences between wild-derived strains and classical inbred strains, which are primarily descended from domesticated mice(10.1038/nature06067),(10.1038/ng2087),(10.1038/ng.847). Additionally, among strains, there is variability in the degree to which particular genomic regions are shared owing to the complex breeding history. Traditional association statistical tests make the assumption that the phenotypes of individuals in an association are independent. However, owing to the complex genetic relationships, this assumption is violated for mouse GWASs. Closely related strains will have more similar phenotype values than more distant strains. This phenomenon, which is termed population structure, causes spurious associations in GWASs. Recently, statistical methods have been developed to address this problem, including efficient mixed-model association (EMMA)(18385116) and resample model averaging (RMA)(10.1534/genetics.109.100727), which are widely used in mouse GWASs, and EIGENSTRAT(10.1038/ng1847) and EMMAX(20208533), which are widely used in human studies. The figure demonstrates this problem for mouse GWASs. Panel a shows body-weight data for 38 inbred strains from the Mouse Phenome Database as analysed in Kang et al., (2008) (18385116). A phylogeny of the strains is shown, demonstrating a clear genetic distinction between the wild-derived strains and the classical inbred strains. Note that all wild-derived strains have a lower body weight than classical inbred strains. Panel b shows a Manhattan plot with the association results for 140,000 SNPs(20439770) and body weight. Almost every locus appears to be associated with body weight as each of the many SNPs that differentiate the wild-derived and classical inbred strains appears to be associated with body weight. A visualization of the cause of the spurious associations is shown panel c. Many SNPs and the phenotype are both correlated with the genetic relatedness or population structure among the strains. Statistical techniques can take into account the genetic relationships between the strains to correct for population structure, thus minimizing spurious associations. In this example, EMMA was applied to the data (panel d). The highest peak, although not genome-wide significant, occurs on chromosome 8 and is near the logarithm of the odds (lod) peak of a previously known body weight quantitative trait locus Bwq3(11515095). Panels b and d are reproduced, with permission, from Kang et al., (2008) (18385116) © (2008) Genetics Society of America.

Pingback: All Excited About Mixed Models | Our 2 SNPs…®
Pingback: ZarLab · Correcting Population Structure using Mixed Models Webcast