
Computational Genomics Summer Institute
CGSI is a unique opportunity for scholars to foster relationships and unleash the full potential of their projects.

UCLA Computational Genetics



Dat Duong, a graduate student in our lab, developed a novel method that will help find more eQTLs and eGenes in gene expression data from many tissues. A paper presenting his method is published in an upcoming issue of Bioinformatics.
Genome-wide association studies (GWAS) seek links between single-nucleotide polymorphisms (SNPs) and traits or diseases. SNPs are the most commonly occurring sources of variation in the human genome. Many SNPs identified by GWAS are located in intergenic regions, stretches of DNA sequences located between genes. SNPs identified in these primarily noncoding regions often do not have an obvious relationship to the disease phenotype. Other lines of evidence, such as gene expression, are required to explore this relationship and learn about disease function.
Gene expression, an intermediate phenotype between a causal SNP and a disease, can be used to interpret positive results produced by a GWAS. Common data types include expression quantitative trait loci (eQTLs), genetic variants associated with gene expression in particular tissue types, and eGenes, genes whose expression levels are associated with genetic variants. Both eQTL studies and GWAS focus on SNPs, but eQTL studies may provide biological insights into the disease development mechanism. For this reason, we pay special attention to the variants that are eQTLs or eGenes and have strong association signals identified by GWAS.
Multi-tissue gene expression datasets like the Gene Tissue Expression (GTEx) data are used to find eQTLs and eGenes. However, these datasets have small sample sizes in some tissues. Many meta-analysis methods have been designed to increase power for finding eQTLs and eGenes by combining gene expression data across many tissues However, these techniques cannot scale to datasets containing many tissue types, like the GTEx data. Such methods also ignore a biological principle that the same variant may be associated with the same gene across similar tissues.
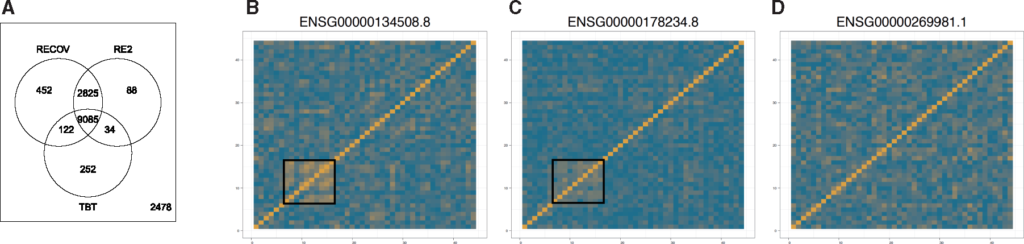
Venn diagram of the numbers of eGenes found by existing methods and RECOV, along with correlation matrices comparing methods. For more information, read our full paper.
To leverage the analytical power of eQTLs and eGenes in association studies, Duong and his team developed a new meta-analysis method named RECOV. Based on the principle that a SNP may have similar effect on the same gene in related tissues, RECOV can be applied to large gene expression datasets and can analyze all 44 tissues present in the GTEx data.
In our Bioinformatics paper, we use simulated datasets to show that RECOV has a correct false positive rate. When applied to real multi-tissue expression data from the GTEx dataset, RECOV detects 3% more eGenes than previous methods. RECOV is a general framework for meta-analysis that can be used with any COV matrix. We hope this software will be used by other researchers in the scientific community!
RECOV was developed by Dat Duong. The source code for RECOV is freely available at: https://github.com/datduong/RECOV.
Our paper can be downloaded at Bioinformatics: https://academic.oup.com/bioinformatics/article/33/14/i67/3953939/Applying-meta-analysis-to-genotype-tissue
The full reference for our paper is:
Duong, D., Gai, L., Snir, S., Kang, E.Y., Han, B., Sul, J.H. and Eskin, E., 2017. Applying meta-analysis to Genotype-Tissue Expression data from multiple tissues to identify eQTLs and increase the number of eGenes. Bioinformatics, 33(14), pp.i67-i74.

Serghei Mangul and Lana Martin, together with Alexander Hoffmann, Matteo Pellegrini, and Eleazar Eskin, recently published a paper describing a workshop model for training scientists, who have no computer science background, to use UNIX. Our paper is available online as a preprint and will appear in an upcoming “Scientific Life” section of Trends in Biotechnology.
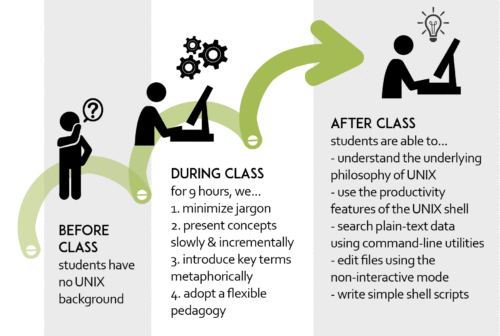
Scientists who are not trained in computer science face an enormous challenge analyzing high-throughput data. Serghei developed a series of workshops in response to growing demand for life and medical science researchers to analyze their own data using the command line.
Administered by UCLA’s Institute for Quantitative and Computational Biosciences (QCBio), these workshops are designed to help life and medical science researchers use applications that lack a graphical interface. Our paper presents a training model for these workshops—a flexible approach that can be implemented at any institution to teach use of command-line tools when the learner has little to no prior knowledge of UNIX.
QCBio currently offers similar workshops to the UCLA community. In tandem with this publication, we created an online catalogue of resources and papers aimed to provide first-time learners with basic knowledge of command line: https://smangul1.github.io/command-line-teaching/.
We encourage fellow instructors of Bioinformatics, as well as scientists who are new learners of the command line, to read our paper and share their thoughts! Email us at: lana [dot] martin [at] ucla [dot] edu.
The full citation of our paper:
Mangul, Serghei, Martin, Lana S., Hoffmann, Alexander, Pellegrini, Matteo, and Eskin, Eleazar. Addressing the Digital Divide in Contemporary Biology: Lessons from Teaching UNIX. Trends in Biotechnology; doi: 10.1016/j.tibtech.2017.06.007.
Advance preprint copies of our paper may be downloaded here: http://www.cell.com/trends/biotechnology/fulltext/S0167-7799(17)30156-7

On Wednesday, June 7, faculty, staff, and undergraduates began a new tradition for the UCLA Bioinformatics Minor Program. Prof. Eleazar Eskin, chair of the Minor Program, hosted the first annual Bioinformatics Minor End-of-Year Celebration to recognize and celebrate undergraduates who completed the requirements for the Minor.
This year’s celebration took place in the Hacienda Room at the Faculty Center. In attendance were faculty and staff from the Schools of Medicine, Engineering, and Life Sciences, and from the Office of Academic and Student Affairs. The reception kicked off with a poster session presenting research completed during the Minor program. As a founding faculty member of the Minor, Prof. Eskin shared a few remarks on the small-scale origins—and upward-scaling trajectory—of the Minor Program. Since 2012, the Minor has succeeded at providing non-computational students with a solid foundation in, and familiarity with, active research problems at the interface of computer science, biology, and mathematics.
Greg Darnell, a UCLA alumnus who completed the Bioinformatics Minor in 2013 and is now a PhD student in Bioinformatics at Princeton, delivered a keynote speech. Mr. Darnell emphasized the extent to which the field of Bioinformatics has changed since he began coursework and research as an undergraduate at UCLA in the early 2010s. Recent “big data” explosions in the Biosciences have created exciting challenges and opportunities for emerging scholars—such as the graduates honored at this reception.
Twenty students were awarded certificates of recognition for their intellectual curiosity, creativity and dedication to interdisciplinary studies. These students join a unique and exemplary group of Bioinformatics Minor graduates who have made their mark in one of UCLA’s most challenging and unique academic programs, spanning Engineering, Biology, and Medicine.
In addition, Prof. Eskin presented three cash research awards to students, Brandon Jew, Ruth Johnson, and Michael Thompson, who demonstrated exceptional talent in their Bioinformatics research projects. Faculty and staff associated with the UCLA Bioinformatics Minor Program are excited to continue this new tradition, including the awarding of certificates and research awards, in future Spring quarters.
Congratulations to the 2016-17 graduates and graduating seniors in the Bioinformatics Minor! In addition to this year’s graduates, a certificate of recognition was also presented to past graduates Leah Briscoe (‘16), Alec Chiu (’16), and Greg Darnell (‘13). Go Bruins!
2016-17 Bioinformatics Minor Graduates:
Ariane Ayer
Andrea Castro
Elizabeth Chin
Qing Dai
Crystal Han
Brandon Jew
Ruth Johnson
Maegan Lu
Anastasia Lukianchikov
Cristian Medina
Douglas Meyer
Sepideh Parhami
Xingyi Shi
Christian Garrison St Pierre
Michael Thompson
Neerja Vashist
Linqing Wei
Anthony Bohr Zhu
See other blog posts on undergraduate training at UCLA:


