
Computational Genomics Summer Institute
CGSI is a unique opportunity for scholars to foster relationships and unleash the full potential of their projects.

UCLA Computational Genetics



Last week many members of our group traveled to Hong Kong, China, for the 21st Annual International Conference on Research in Computational Molecular Biology (RECOMB 2017). This year’s meeting, which took place May 3-7, 2017, featured over 300 talks, workshops, and poster presentations on topics from all areas of computational molecular biology.
This year, our group contributed 7 poster presentations, 2 research talks, and 1 research highlight (see below for a complete list). In addition to science, ZarLab members enjoyed traveling about Hong Kong and taking in the food, sights, and bicycle paths.
Our Papers:
Yue (Ariel) Wu, Farhad Hormozdiari, Jong Wha J Joo and Eleazar Eskin.
Improving imputation accuracy by inferring causal variants in genetic studies
https://link.springer.com/chapter/10.1007/978-3-319-56970-3_19
Elior Rahmani, Regev Schweiger, Liat Shenhav, Eleazar Eskin and Eran Halperin.
A Bayesian Framework for Estimating Cell Type Composition from DNA Methylation Without the Need for Methylation Reference
https://link.springer.com/chapter/10.1007/978-3-319-56970-3_13
Our Highlights:
Elior Rahmani, Noah Zaitlen, Yael Baran, Celeste Eng, Donglei Hu, Joshua Galanter, Sam Oh, Esteban Burchard, Eleazar Eskin, James Zou and Eran Halperin
Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies
Our Posters:
Yue (Ariel) Wu, Eleazar Eskin and Sriram Sankararaman
Improving imputation by maximizing power
Lisa Gai, Dat Duong and Eleazar Eskin
Finding associated variants in genome-wide associations studies on multiple traits
Harry Taegyun Yang, Serghei Mangul, Noah Zaitlen, Sagiv Shifman and Eleazar Eskin
Repeat Elements Profile Across Different Tissues in GTEx Samples
Dat Duong, Lisa Gai, Sagi Snir, Eun Yong Kang, Buhm Han, Jae Hoon Sul and Eleazar Eskin
Applying meta-analysis to Genotype-Tissue Expression data from multiple tissues to identify eQTLs and increase the number of eGenes
Jennifer Zou, Farhad Hormozdiari, Jason Ernst, Jae-Hoon Sul and Eleazar Eskin
Leveraging allele-specific expression to improve fine-mapping for eQTL studies
Serghei Mangul, Igor Mandric, Alex Zelikovsky and Eleazar Eskin
Profiling adaptive immune repertoires across multiple human tissues by RNA Sequencing
Robert Brown, Eleazar Eskin and Bogdan Pasaniuc
Haplotype-based eQTL Mapping Increases Power to Identify eGenes

This week, our group published a paper in the American Journal of Human Genetics that presents a new computational method for improving the accuracy of genome wide association studies. ZarLab alumni Farhad Hormozdiari (PhD, 2016) developed the method, CAVIAR (CAusal Variants Identification in Associated Regions), a statistical framework that quantifies the probability of each variant to be causal while allowing an arbitrary number of causal variants.
Genome-wide association studies (GWASs) identify genetic variants associated with diseases and traits. Recent successes in GWASs make it possible to address important questions about the genetic architecture of complex traits, such as allele frequency and effect size. A more comprehensive understanding of these aspects will guide the development of new methods for fine mapping and association mapping of complex traits—and the discovery of new biomarkers for disease diagnosis and treatment.
One lesser-known aspect of complex traits is the extent of allelic heterogeneity (AH). Allelic heterogeneity occurs when different mutations at the same locus affects the same phenotype. AH is very common in Mendelian traits, but we know little about the extent to which AH contributes to common, complex disease. Undetected AH could potentially bias results of an association study, leading to false positive results.
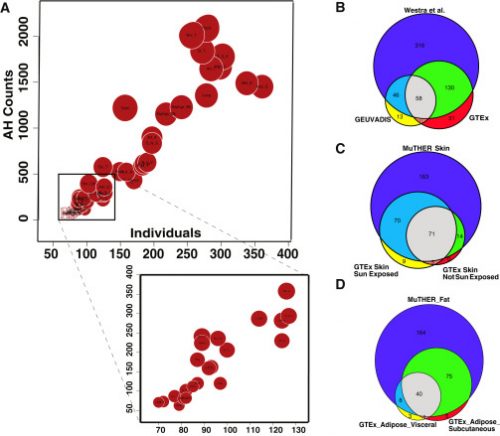
Levels of Allelic Heterogeneity in eQTL Studies. For more information, see our paper.
In order to take AH into account while conducting a GWAS, we developed a computational method to infer the probability of AH. Our method quantifies the number of independent causal variants at a locus that can be responsible for the observed association signals detected in a GWAS. Our method is incorporated into the CAVIAR approach, and it is based on the principle of jointly analyzing association signals (i.e., summary level Z-scores) and LD structure in order to estimate the number of causal variants.
Our results show that our method is more accurate than the standard conditional method (CM). We applied our novel method to three GWASs and four expression quantitative trait loci (eQTL) datasets. We identified a total of 4,152 loci with strong evidence of the presence of AH. The proportion of all loci with identified AH is 4%–23% in eQTLs, 35% in GWASs of high-density lipoprotein (HDL), and 23% in GWASs of schizophrenia. For eQTLs, we observed a strong correlation between sample size and the proportion of loci with AH, indicating that statistical power prevents identification of AH in other loci.
One of the main benefits of our method is that it requires only summary statistics. Summary statistics of a GWAS or eQTL study are widely available, so our method is applicable to most existing datasets. We have shown that AH is widespread and more common than previously estimated in complex traits, both in GWASs and eQTL studies.
Our results highlight the importance of accounting for the presence of multiple causal variants when characterizing the mechanism of genetic association in complex traits. Falling to account for AH can reduce the power to detect true causal variants and can explain the limited success of fine mapping of GWASs.
In a related study, researchers at University of California, Irvine, and University of Kansas, identified an analogous signal in eQTLs from genetic sequencing of flies. King et al. (2014) observe that the vast majority of genes with eQTL are more consistent with heterogeneity than bi-allelism. Read more about this related study, “Genetic Dissection of the Drosophila melanogaster Female Head Transcriptome Reveals Widespread Allelic Heterogeneity.”
CAVIAR was created by Farhad Hormozdiari, Emrah Kostem, Eun Yong Kang, Bogdan Pasaniuc and Eleazar Eskin. Software is freely available for download: http://genetics.cs.ucla.edu/caviar/
For more information, see our full paper, which can be accessed through AJHG: http://www.cell.com/ajhg/abstract/S0002-9297(17)30149-0
The full citation of our paper:
Hormozdiari F, Zhu A, Kichaev G, Ju CJ, Segrè AV, Joo JW, Won H, Sankararaman S, Pasaniuc B, Shifman S, Eskin E. Widespread allelic heterogeneity in complex traits. The American Journal of Human Genetics. 2017 May 4;100(5):789-802.
Our group, in an effort led by former UCLA PhD student Dan He, developed an algorithm for reconstructing pedigrees with genotype data. This novel approach is presented in a paper recently published in IEEE/ACM Transactions on Computational Biology and Bioinformatics.
Pedigree inference plays an important role in population genetics. Pedigrees, commonly known as family trees, represent genetic relationships between individuals of a family. A pedigree diagram provides a model to compute the inheritance probability for the observed genotype and encodes all possible inheritance options for an allele in an individual. Pedigree reconstruction methods face several challenges. First, there can be an exponential number of possible pedigree graphs, and, second, the number of unknown ancestors can become very large as the height of the pedigree increases.
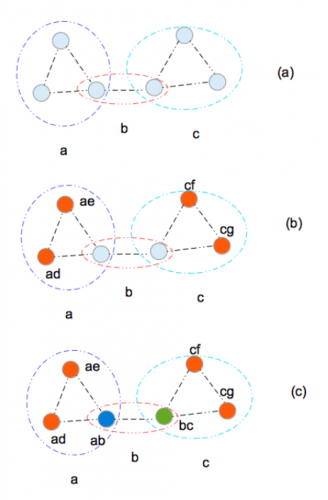
Examples of sequentially labeling the half-sibling graph. For more information, see our paper.
Our project uses genotype data to reconstruct pedigrees with computational efficiency despite these challenges. Our previous method, IPED, is the only known algorithm scalable to large pedigrees with reasonable accuracy for cases involving both outbreeding and inbreeding. IPED starts from extant individuals and reconstructs the pedigree generation by generation backwards in time. For each generation, IPED predicts the pairwise relationships between the individuals at the current generation and create parents for them according to their relationships.
Existing methods, including IPED, only consider pedigrees with simple structure; they cannot handle populations where, for example, two children share only one parent. To improve pedigree reconstruction when populations have complex structure, we proposed the novel method IPED2. Our approach uses a new statistical test to detect half-sibling relationships and a new graph-based algorithm to reconstruct the pedigree when half-siblings are allowed.
In order to test the performance of our method on complicated pedigrees, we use simulated pedigrees with different parameter settings and, instead of genotype data, we simulate haplotypes
directly. Our experiments show that IPED2 outperforms IPED and two other existing approaches for cases where there are half-siblings.
To our knowledge, this is the first method that can, using just genotype data, reconstruct pedigrees with half-siblings and inbreeding. IPED2 is also scalable to large pedigrees. In future work, we would like to consider additional genetic actions, such as insertion, deletion, and replacement, to resolve the conflicts. We also plan to refine IPED2 to consider cases where genotypes of ancestral individuals are known and where genotypes of extant individuals that are not on the lowest generations are known.
For more information, see our paper, which is available for download through Bioinformatics: http://ieeexplore.ieee.org/abstract/document/7888513/.
In addition, the open source implementation of IPED2, which was developed by Dan He, is freely available for download at http://genetics.cs.ucla.edu/Dan/Software/IPED2.html.
The full citation to our paper is:
He, D., Wang, Z., Parida, L. and Eskin, E., 2017. IPED2: Inheritance path based pedigree reconstruction algorithm for complicated pedigrees. IEEE/ACM Transactions on Computational Biology and Bioinformatics.


