
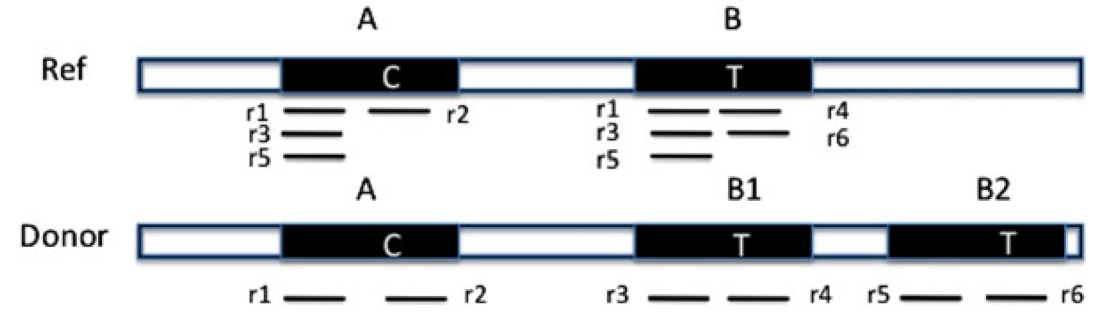
Similar copies of a copy number variations (CNV) region exist in the reference genome. ‘‘C’’ and ‘‘T’’ are the only different nucleotides between region A and B. Reads {r1‚r2‚…‚r6} are obtained from the donor genome as shown in the lower part of the figure. Furthermore, these reads can be mapped to the reference genome as shown in the upper part of the figure.
Identifying copy number variation from high throughput sequencing data is a very active research area(10.1038/nrg2958). Typical approaches map short sequence reads from a donor genome to a reference genome and then examine the number of reads that map to each region. The idea is that if few reads map to a region, this suggests that the corresponding portion of the donor genome was deleted and a large number of reads mapping to a region suggests that the corresponding region is duplicated in the donor genome.
This method works very well when the duplicated or deleted region is unique and reads originating from that region can only map to a single location. Unfortunately, many copy number variations occur in regions which themselves are duplicated in the genome. Reads originating from these regions map to multiple positions in the reference. Incorrect placement of these reads can then result in wildly incorrect copy number predictions.
We recently published a paper on dealing with read mapping uncertainty when predicting copy number variation(10.1089/cmb.2012.0258). Instead of mapping the reads to a single location, we keep a probability distribution over all of the locations that they can map. Then we iteratively estimate the copy number and then remap the reads using these estimates. What results is that the few reads that span the small number of differences between the copies (as shown in the figure from the paper) end up being the clues to correctly determine which region was copied.
Ph.D. students Zhanyong Wang, Farhad Hormozdiari, Wen-Yun Yang worked on this project which was a collaboration with Eran Halperin.
Full Citation:
Wang, Zhanyong, Farhad Hormozdiari, Wen-Yun Yang, Eran Halperin, and Eleazar Eskin. 2013. CNVeM: Copy number variation detection using uncertainty of read mapping. J Comput Biol doi:10.1089/cmb.2012.0258
Abstract:
Copy number variations (CNVs) are widely known to be an important mediator for diseases and traits. The development of high-throughput sequencing (HTS) technologies has provided great opportunities to identify CNV regions in mammalian genomes. In a typical experiment, millions of short reads obtained from a genome of interest are mapped to a reference genome. The mapping information can be used to identify CNV regions. One important challenge in analyzing the mapping information is the large fraction of reads that can be mapped to multiple positions. Most existing methods either only consider reads that can be uniquely mapped to the reference genome or randomly place a read to one of its mapping positions. Therefore, these methods have low power to detect CNVs located within repeated sequences. In this study, we propose a probabilistic model, CNVeM, that utilizes the inherent uncertainty of read mapping. We use maximum likelihood to estimate locations and copy numbers of copied regions and implement an expectation-maximization (EM) algorithm. One important contribution of our model is that we can distinguish between regions in the reference genome that differ from each other by as little as 0.1%. As our model aims to predict the copy number of each nucleotide, we can predict the CNV boundaries with high resolution. We apply our method to simulated datasets and achieve higher accuracy compared to CNVnator. Moreover, we apply our method to real data from which we detected known CNVs. To our knowledge, this is the first attempt to predict CNVs at nucleotide resolution and to utilize uncertainty of read mapping.
