
Farhad Hormozdiari recently developed a method for combining genome-wide association studies (GWASs) and quantitative trait loci (eQTL) studies in a statistical framework that quantifies the probability of each variant to be causal while allowing an arbitrary number of causal variants. Together with collaborators at the University of Oxford and Broad Institute of MIT and Harvard, we present a paper in The American Journal of Human Genetics. Here, we describe eQTL and GWAS CAusal Variants Identification in Associated Regions (eCAVIAR). We apply our approach to datasets from several GWASs and eQTL studies in order to assess its accuracy and potential contributions to colocalization and fine-mapping.
Integrating GWASs and eQTL studies is a promising way to explore the mechanism of non-coding variants on diseases. Integration of GWAS and eQTL data is challenging due to the uncertainty induced by linkage disequilibrium (LD), the non-random association of alleles at different loci, and presence of loci that harbor multiple causal variants (allelic heterogeneity). Current methods assume that each locus contains a single causal variant and expect loci to be independent and associated randomly.
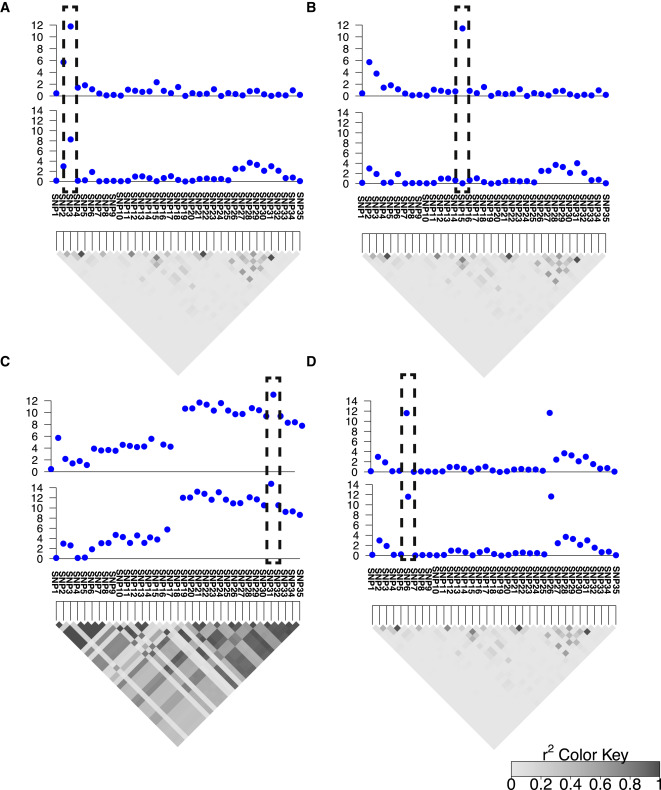
eCAVIAR is a novel probabilistic model for integrating GWAS and eQTL data that extends the CAVIAR (Hormozdiari et al. 2014) framework to explicitly estimate the posterior probability of the same variant being causal in both GWAS and eQTL studies, while accounting for allelic heterogeneity and LD. Our approach can quantify the strength between a causal variant and its associated signals in both studies, and it can be used to colocalize variants that pass the genome-wide significance threshold in GWAS. For any given peak variant identified in GWAS, eCAVIAR considers a collection of variants around that peak variant as one single locus.
We apply eCAVIAR to the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) dataset and GTEx dataset to detect the target gene and most relevant tissue for each GWAS risk locus. When applied to the MAGIC dataset’s 2 phenotypes, eCAVIAR identifies genetic variants that are causal in both eQTL and GWAS. Further, eCAVIAR detects a large number of loci where the GWAS causal variants are clearly distinct from the causal variants in the eQTL data. Interestingly, eCAVIAR also identifies genes that colocalize in one tissue yet can be excluded in others. For the majority of loci in which we identify a single variant causal for both GWAS and eQTL, eCAVIAR implicates more than one causal variant across the 45 tissues.
We observe that eCAVIAR outperforms existing methods even when there are different values of non-colocalization. Using simulated datasets, we compared accuracy, precision, and recall rate of eCAVIAR to RTC (Nica et al. 2010) and COLOC (Giambartolomei et al. 2014), two current methods for eQTL and GWAS colocalization. Our results show that eCAVIAR has high confidence for selecting loci to be colocalized between the GWAS and eQTL data and is conservative in selecting a locus to be colocalized.
We hope that future applications of eCAVIAR will advance identification of specific GWAS loci that share a causal variant with eQTL studies in a tissue, thus providing insight into presently unclear disease mechanisms.
Overview of eCAVIAR.
eCAVIAR was created by Farhad Hormozdiari, Ayellet V. Segre, Martijn van de Bunt, Xiao Li, Jong Wha J Joo, Michael Bilow, Jae Hoon Sul, Bogdan Pasaniuc and Eleazar Eskin. The article is available at: http://www.cell.com/ajhg/abstract/S0002-9297(16)30439-6.
Visit the following page to download CAVIAR and eCAVIAR: http://genetics.cs.ucla.edu/caviar/
The full citation to our paper is:
Our paper builds upon a method introduced in a previous publication:
