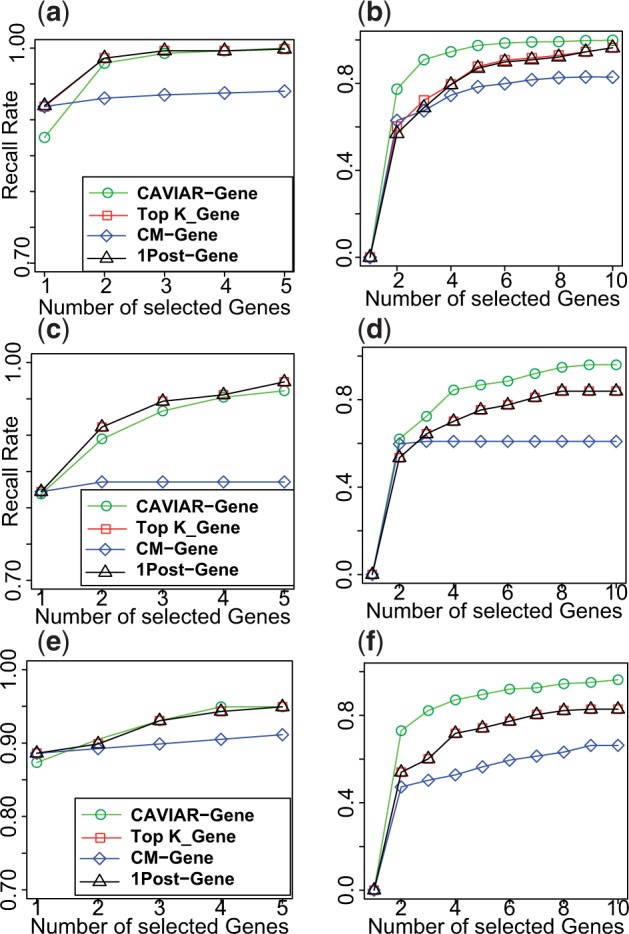
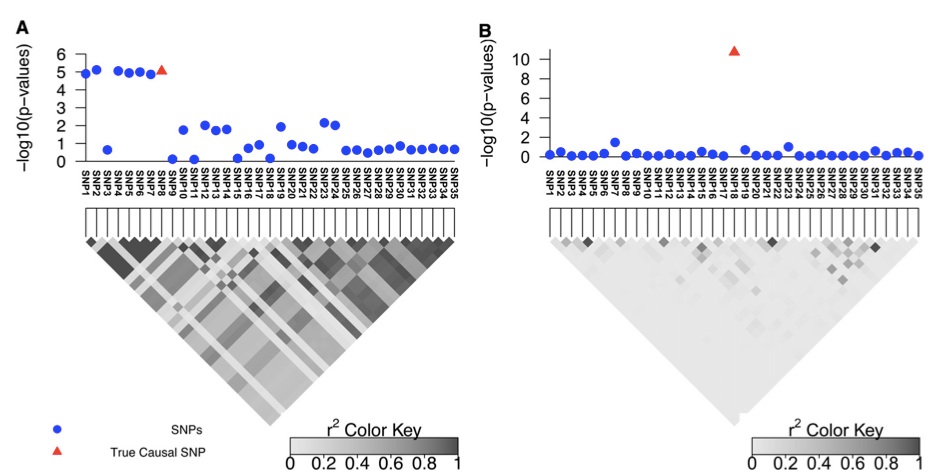
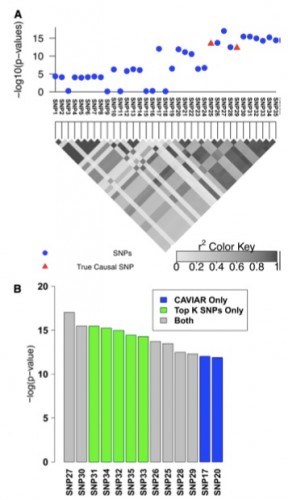
Farhad Hormozdiari, a recent ZarLab alumni, contributed to a paper published this week in Nature. Our paper reports new findings on genetic factors related to human cognition and neurodevelopmental disorders, the result of a collaboration with UCLA’s David Geffen School of Medicine and the School of Biotechnology and Biomolecular Sciences at University of New South Wales. Farhad implemented the software package CAVIAR which was utilized to identify the causal variants and interpretation of data.
Neurodevelopmental disorders such as autism and schizophrenia are thought to originate during embryonic development of the cerebral cortex. The project focused on the 3D interactions of genome-wide chromatin contacts, the areas of a cell’s nucleus that package chromosomes into DNA and influence cell replication. Chromatin contacts regulate gene expression in specific tissues, and mapping their interactions within chromosomes provides important biological insights into the malfunctioning gene regulatory mechanisms that drive these disorders.
The project generated high-resolution 3D maps of chromatin contacts active during development of the cortex region of the human brain. These maps enabled a large-scale annotation of previously uncharacterized regulatory mechanisms tied to the evolution of human cognition and disease. Using this data, the paper identified hundreds of genes involved with human cognitive function. Next, the paper integrated chromatin contacts with noncoding variants previously identified in schizophrenia genome-wide association studies (GWAS) and performed several analyses to explore the relationships of interactions between chromatin and biological function. One of the uses of CAVIAR in the paper was to verify that the causal variants involved in schizophrenia GWAS are in fact compatible with the 3D maps of chromatin contacts.
The paper also found several highly interacting chromatin regions that correlate with levels of gene expression and are associated with promoters, positive transcriptional regulators, and enhances—areas of the genome that shape cell replication and neurological development. The paper identified specific sets of genes enriched in known intellectual disability risk genes, including mutations known to cause autosomal recessive primary microcephaly. The GWAS results identified approximately 500 genome-wide significant schizophrenia-associated loci, about 30% of which interact with schizophrenia SNPs exclusively in developing brain tissue. Genome editing in human neural progenitors suggests that one of these distal schizophrenia GWAS loci regulates FOXG1 expression, supporting its potential role as a schizophrenia risk gene.
This work provides a framework for understanding the effect of non-coding regulatory elements on human brain development and the evolution of cognition, and highlights novel mechanisms underlying neuropsychiatric disorders. Read the paper for a detailed account of our data, methods, and results: http://www.nature.com/nature/journal/vaop/ncurrent/full/nature19847.html
The CAVIAR program was developed by Farhad Hormozdiari and is freely available for download on the following webpage: http://genetics.cs.ucla.edu/caviar/
The full citation to our paper is:
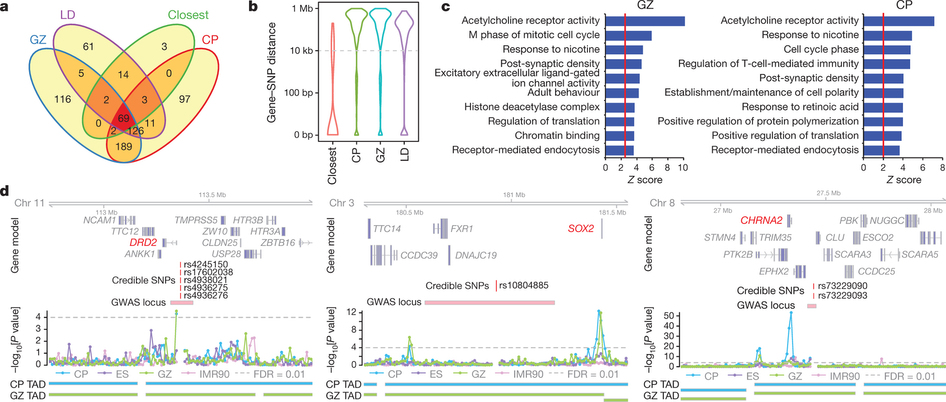
Annotation of schizophrenia-associated loci identified by a GWAS of chromatin contact data.