Farhad Hormozdiari and Eleazar Eskin recently applied an extension of CAVIAR to assess signal selection in European ancestry. CAVIAR is a probabilistic method for detecting a confidence set of SNPs containing all the causal variants in a locus that are within a predefined probability (e.g., 90% or 95%)—while taking into account biases generated by linkage disequilibrium. Farhad, now a post-doctoral scholar at Boston University, developed CAVIAR while a PhD student at UCLA.
This project was led by Matthew T. Buckley and Fernando Racimo at the University of California, Berkeley, and Morten E. Allentoft at the University of Copenhagen. Alleles with strong selection signals have been recently selected for and are thought to carry an evolutionary advantage for individuals in the population. Identifying these alleles helps expand our understanding of the selective pressures that shaped historic populations.
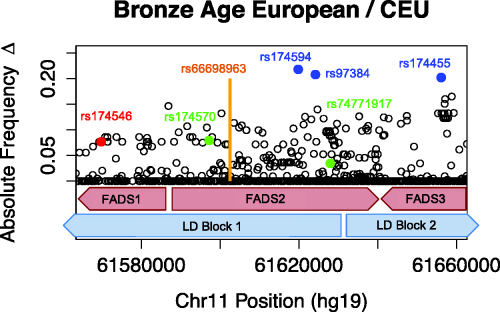
Allele frequency changes across FADS region. For more information, see our full paper.
In order to analyze the selective processes in Europeans across space and time, the project compared sequencing data from FADS genes obtained from present-day and Bronze Age (5000 to 3000 years ago) Europeans. We focused on FADS genes because prior studies indicate they are subjected to strong positive selection in Africa, South Asia, Greenland, and Europe. FADS genes encode fatty acid desaturases that are important for the conversion of short chain polyunsaturated fatty acids (PUFAs) to long chain fatty acids. In other words, selective pressure in the FADS genes may be linked to dietary adaptations.
Other analyses conducted by the project show that alleles in the FAD2 gene display the strongest changes in allele frequency since the Bronze Age, and this change shows associations with expression changes and multiple lipid-related phenotypes. Farhad and Eleazar used CAVIAR to look for presence of allelic heterogeneity, an adaptive process in which different mutations at the same locus cause the same phenotype. In an evolutionary context, presence suggests that a strong pressure selective pressure likely acted upon the population.
Application of CAVIAR to genomic data from the 1000 Genomes Project and 54 Bronze Age Europeans revealed that specific causal variants within the FADS2 gene have been subjected to selective pressure. In particular, FADS2 shows evidence of allelic heterogeneity in three tissue types: transformed fibroblast cells (Pr(2 causal variants) = 0.72), left heart ventricle (Pr(2 causal variants) = 0.74), and whole blood (Pr(3 causal variants) = 0.74).
The project’s comparison of modern to Bronze Age European genomic data show that selection has indeed strongly acted on the FADS gene cluster over the past 3000 years. The selective patterns observed in European data may be driven by a change in the dietary composition of fatty acids following the human transition from hunting-and-gathering to agriculture. As Europeans obtained more lipids from plants, rather than from fish and mammals, their genes adapted to optimize metabolism of these cereal-based lipids.
For more information, see our paper, which is available for download through Molecular Biology and Evolution: https://www.ncbi.nlm.nih.gov/pubmed/28333262.
The full citation to our paper is:
Buckley, M.T., Racimo, F., Allentoft, M.E., Jensen, M.K., Jonsson, A., Huang, H., Hormozdiari, F., Sikora, M., Marnetto, D., Eskin, E. and Jørgensen, M.E., 2017. Selection in Europeans on fatty acid desaturases associated with dietary changes. Molecular biology and evolution.
This project used a method introduced in a previous publication:
CAVIAR was created by Farhad Hormozdiari, Emrah Kostem, Eun Yong Kang, Bogdan Pasaniuc, and Eleazar Eskin. Visit the following page to download CAVIAR and eCAVIAR: http://genetics.cs.ucla.edu/caviar/.