Genome-wide association studies (GWAS) seek to identify genetic variants involved in specific traits. GWAS are advantageous for linking variants with traits, because they interrogate the genome in a uniform way. In other words, they examine the whole genome without a preconceived notion of where the associations may lie.
However, we now know a lot about the putative function of genetic variants due to tremendous progress in functional genomics. In many cases, we even know which variants are more likely to be involved in disease when compared to others. Advancements in our understanding of functional genomics motivate the strategic incorporation of prior information in GWAS.
Our group has been interested in this problem for many years. One challenge to addressing this problem is that the widely utilized approach for GWAS involves evaluating an association statistic at each single nucleotide polymorphism (SNP), and these methods take into account only one SNP at a time. The results are then adjusted for multiple testing, and an association is identified if a statistic exceeds a certain threshold. This approach can be described as a frequentist approach. On the other hand, one can incorporate prior information on which SNPs are likely to be the causal variants affecting the trait. This approach is inherently a Bayesian concept. Reconciling these two approaches is not straightforward.
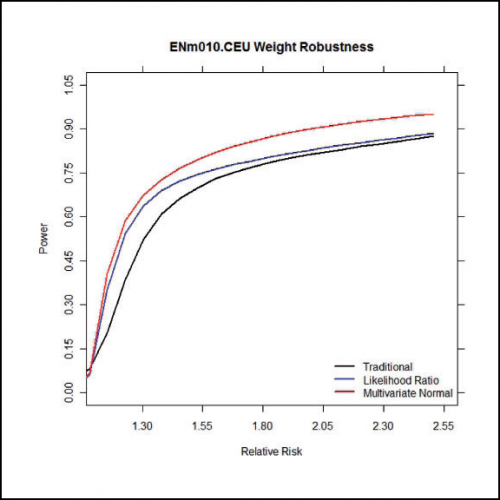
Average power under varying relative risks. For more information, see our paper.
In a 2008 paper published in Genome Research, our group proposed a modification of the multiple testing framework to address this problem. Instead of using the same specific threshold for all of the association statistics, we use a different threshold for each association statistic, where the thresholds are adjusted based on the prior information. Our method takes advantage of the correlation structure by considering multiple markers within a region. In our paper, we demonstrate how to set the thresholds in order to optimally utilize prior information and maximize statistical power.
Using prior information in genetic association studies increases power over traditional association studies while maintaining the same overall false-positive rate. Compared to standard methods, our approach is equally simple to apply to association studies, produces interpretable results as p-values, and is optimal in its use of prior information in regards to statistical power.
In 2012, we extended this work to use only tag SNPs for the putative causal variant. This project was developed by Gregory Darnell (then UCLA undergraduate, now PhD student at Princeton University), Dat Duong (then UCLA undergraduate, now UCLA PhD student), and Buhm Han.
More recently, we have applied this framework to incorporate functional information in analysis of eQTL data. In this case, incorporating genomic annotation of variants significantly increases the statistical power of existing eQTL methods and detects more eGenes in comparison to standard approaches. Read the blog post on this paper, and download the full article.
For more information on our general approach, see our paper, which is available for download through Bioinformatics:
https://academic.oup.com/bioinformatics/article/28/12/i147/269880/Incorporating-prior-information-into-association
In addition, the open source implementation of our 2012 paper, MASA, which was developed by Greg Darnell and Dat Duong, is freely available for download at http://masa.cs.ucla.edu/.
The full citations to our papers on this topic are:
Eleazar Eskin. “Increasing Power in Association Studies by using Linkage Disequilibrium
Structure and Molecular Function as Prior Information.” Genome Research.
18(4):653-60 Special Issue Proceedings of the 12th Annual Conference on Research
in Computational Biology (RECOMB-2008), 2008.