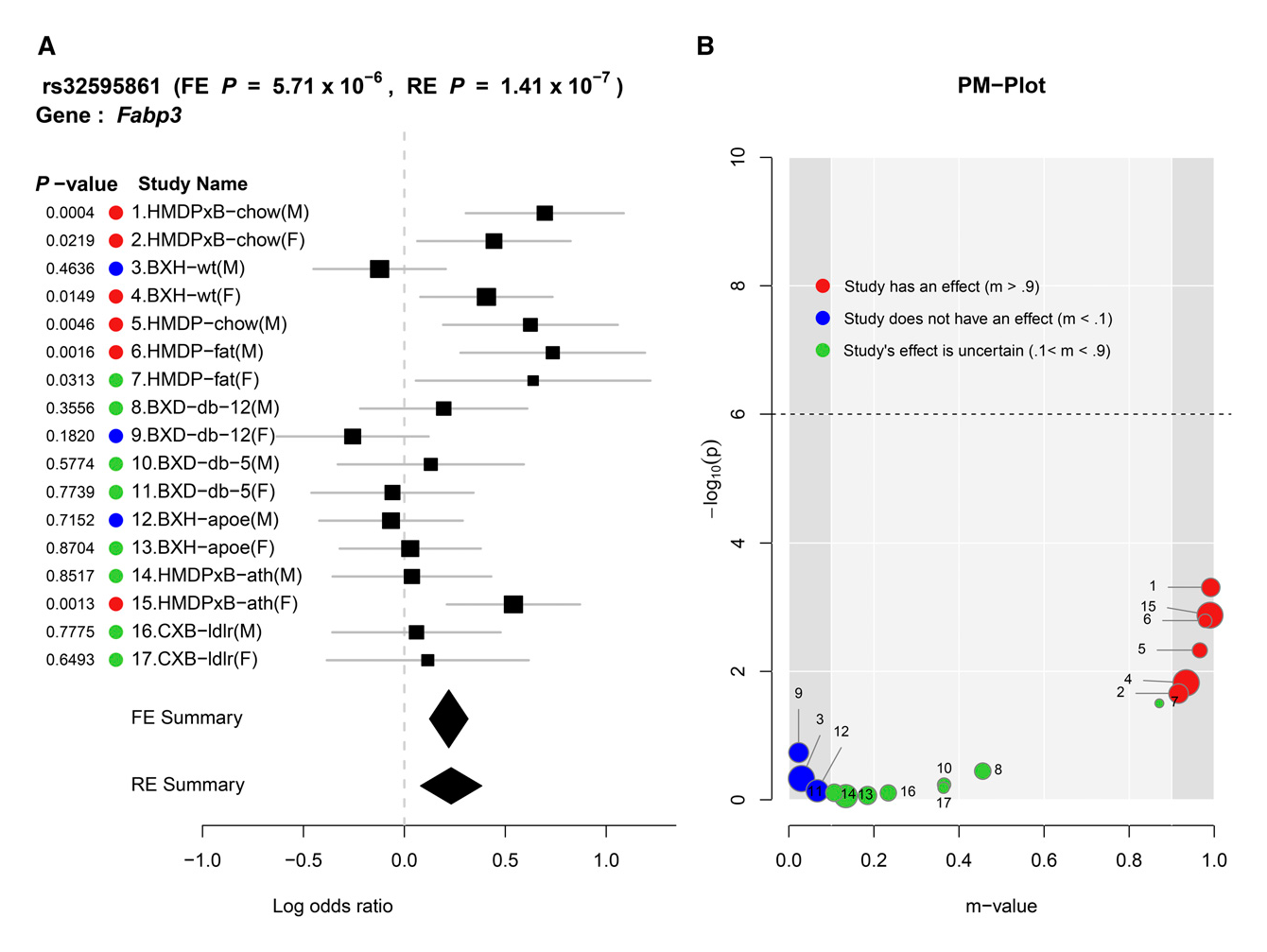
This year, our group published a paper in PLOS Genetics that describes our efforts to better understand and correct for population structure when computing gene-by-environment (GEI) statistics in genome-wide association studies (GWASs). We use simulated and actual GWAS datasets to demonstrate that population structure, the relatedness of individuals within a cohort, inflates test statistics for both GEIs and genetic variants. We present a novel mixed model method capable of improving accuracy when computing GEI statistics in GWAS. This method can be efficiently applied to GWAS datasets containing thousands of individuals and hundreds of thousands of SNPs.
GWASs have discovered many genetic variants associated with complex traits and diseases, yet these genetic variants explain only a small fraction of phenotypic variance in the human genome. Other sources of phenotypic variance include discrete environmental factors and GEIs, complex interactions between an individual’s genetic material and environmental factors. Recent GEI association analyses have demonstrated the importance of GEIs in complex traits and disease development. Identification of these causal GEIs would provide insight into disease pathways, particularly the effects of environmental factors in disease risk, and guide development of novel diagnostic tools and personalized therapies.
Several methodological challenges have limited successful identification of causal GEIs. As with standard GWAS approaches, GxE GWASs are prone to produce an inflated number of associations due to population structure. Unlike standard GWASs, we lack a method designed to avoid detection of these spurious associations when computing GEI statistics. Accounting for genetic similarity with a standard GWAS approach does control inflation of test statistics for causal SNPs, but does not control inflation of associated GEIs. Simultaneously accounting for both similarities would control both types of population structure known to confound GWASs—false associations caused by SNPs under selection and those caused by the remaining SNPs.
Our linear mixed model approach introduces two random effects and takes into account two types of similarities between individuals: overlap in the genome itself and overlap in genetic expression caused by complex interactions between genes and environment. We use a pair of kinship matrices corresponding to the two types of similarity to include these two random effects in the model and correct for population structure.
In order to better understand false associations in GxE GWASs, we compare our approach to two standard approaches. We apply the three methods to two large genomic datasets, one human and one mouse, that are known to contain population structure and have many quantitative phenotypes to test effect of GEIs. We use a standard GWAS method that does not correct for population structure (defined as “OLS” in our paper) and an approach that performs population structure correction for only SNP statistics (“One RE”). The last approach is our proposed mixed model approach that uses both genetic and GxE kinship to correct for population structure on both SNP and GEI statistics (“Two RE”).
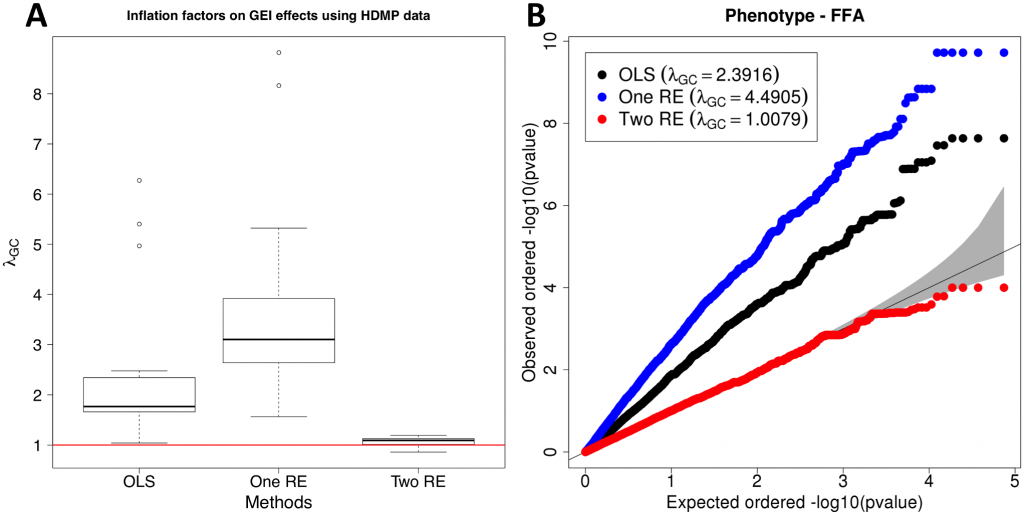
Distribution of inflation factors of GEI statistics on HMDP GxE GWAS data. (A) Inflation factor for each phenotype with no population structure correction (OLS), population structure correction for SNP statistics (One RE), and population structure correction for both SNP and GEI statistics (Two RE). (B) QQ plot of one of the phenotypes (free fatty acids, ffa), showing the distributions of p-values of GEI statistics for the three methods.
In both datasets, even a moderate amount of population structure causes spurious GEIs when using standard approaches for identifying GEI in GWAS. While the One RE approach reduces inflation of test statistics on SNPs (see Supplement S1 Figure), it has almost the same or slightly higher inflation factors on GxE statistics when compared to OLS. Results from both datasets suggest that our approach effectively controls population structure when computing statistics for GEIs and genetic variants. We hope our method is useful advancing our understanding of how life-history influences an individual’s disease risk.
This project was led by Jae Hoon Sul and involved Michael Bilow. The article is available at: http://dx.doi.org/10.1371/journal.pgen.1005849
The full citation to our paper is:
This approach uses our PyLMM software package available for download at: http://genetics.cs.ucla.edu/pylmm/.