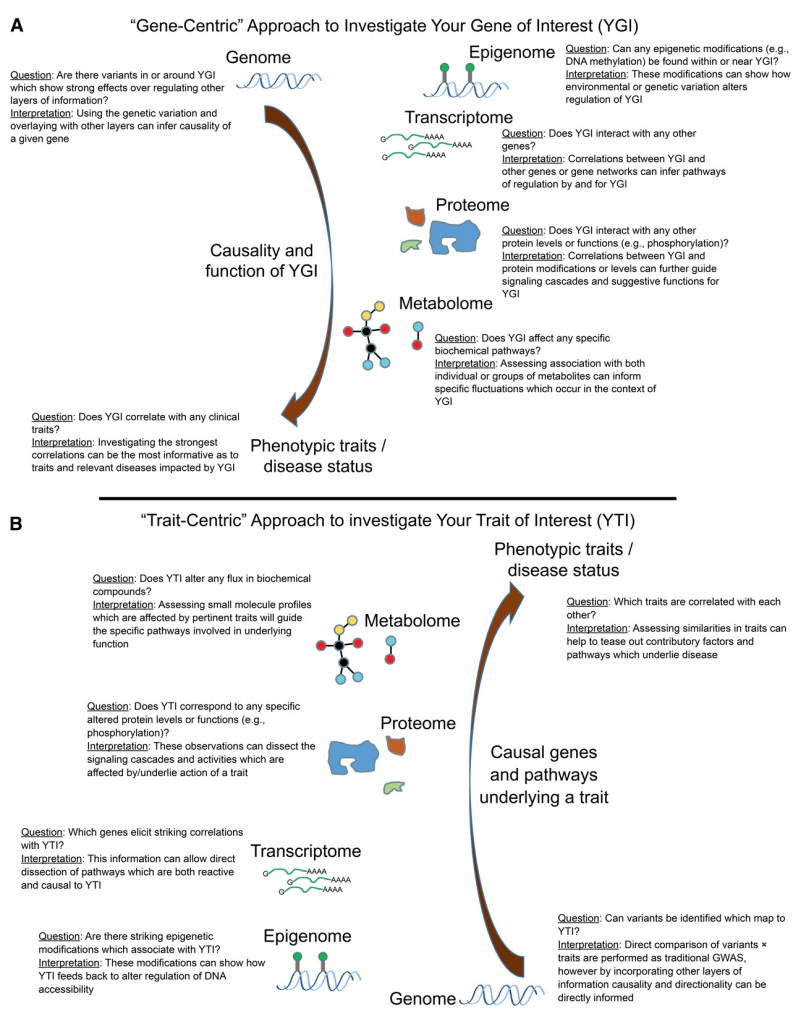
Analyses of expression quantitative trait loci (eQTL), genomic loci that contribute to variation in genetic expression levels, are essential to understanding the mechanisms of human disease. These studies identify regulators of gene expression as either cis-acting factors that regulate nearby genes, or trans-acting factors that affect unlinked genes through various functions. Traditional eQTL studies treat expression as a quantitative trait and associate it with genetic variation. This approach has identified many loci involved in the genetic regulation of common, complex diseases.
Standard eQTL methods are limited in power and accuracy by several phenomena common to genomic datasets. First, the correlation structure of genetic variation in the genome, known as linkage disequilibrium (LD), limits the ability of these methods to differentiate between the regulatory variant and neighboring variants that are in LD. Second, like other quantitative traits, the total expression of a gene is influenced by multiple genetic and environmental factors. The effect size for any given variant is therefore small, and standard methods require a large sample size to identify the effect.
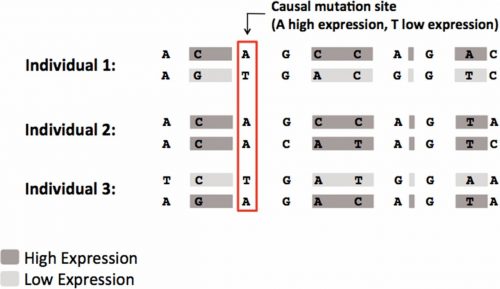
ASE example and corresponding mathematical representation of three individuals (1, 2, 3). We assume that the third SNP is the causal SNP site affecting the differential gene expression level (Allele A/ Allele T).
Our forthcoming paper in Genetics presents a new method that improves the accuracy and computational power of eQTL mapping with incorporation of allele specific expression (ASE) analysis. Our novel method uses genome sequencing, alongside measurements of ASE from RNA-seq data, to identify cis-acting regulatory variants.
In standard eQTLs studies, the analysis of ASE is influenced by LD structure and the amount of allelic heterogeneity present in the genome. Individual effects appear weak since the effect of a variant is modest when compared to the variance of total expression. In our approach, the genotypes of each single individual with ASE provides information useful to determining variants causal for the observed ASE. Our approach actually leverages the relationship between LD and variant identification to map the variants affecting expression. Thus, analysis of ASE is advantageous over analysis of total expression levels, the standard approach to eQTL mapping.
We demonstrate the utility of our method by analyzing RNA-seq data from 77 unrelated northern and western European individuals (CEU). To map each gene, we simultaneously compare ASE measurements across a set of sequenced individuals. We then identify genetic variants that are in proximity to those genes and capable of explaining observed patterns of ASE. Here, we characterize the efficacy of this method as the ratio termed “reduction rate” and denoted as the ratio between the number of candidate regulatory SNPs to the total number of SNPs in the proximal region of the gene.
When applied to the CEU dataset, our method reduced the set of candidate SNPs from ten to two (a reduction rate of 80%). Allowing for one error increases the number of candidate SNPs to five and decreases the reduction rate to 50%. We also observe that the relationship between LD and variant identification has a different quality in ASE mapping when compared to eQTL studies, and produces different types of information useful to eQTL mapping studies.
ASE studies are a powerful approach to identifying associations between genetic variation and gene expression. Accurate measurement of ASE can identify cis-acting regulatory variants associated with common diseases. Our novel method for ASE mapping is based on a robust and computationally efficient non-parametric approach, and we hope it advances our understanding of functional risk alleles and facilitates development of new hypotheses for the causes and treatment of common diseases.
This project used software developed by Jennifer Zou, which is available for download at: http://genetics.cs.ucla.edu/ase/
This project was led by Eun Yong Kang and involved Serghei Mangul, Buhm Han, and Sagiv Shifman. The article is available at: http://www.genetics.org/content/204/3/1057
The full citation to our paper is: