Michael Bilow and Eleazar Eskin, together with Fernando Crespo, Zhicheng Pan, and Susana Eyheramendy, recently released a novel method for accurate joint modeling of clinical phenotype and disease status. This approach incorporates a clinical phenotype into case/control studies under the assumption that the genetic variant can affect both.
Genetic case-control association studies have found thousands of associations between genetic variants and disease. Most studies collect data from individuals with and without disease, and they often search for variants with different frequencies between the groups. Jointly modelling clinical phenotype and disease status is a promising way to increase power to detect true associations between genetics and disease. In particular, this method increases potential for discovering genetic variants that are associated with both a clinical phenotype and a disease.
However, standard multivariate techniques fail to effectively solve this problem because their case-control status is discrete and not continuous. Standard approaches to estimate model parameters are biased due to the ascertainment in case/control studies. We present a novel method that resolves both of these issues for simultaneous association testing of genetic variants that have both case status and a clinical covariate.
In our paper, we show the utility of our method using data from the North Finland Birth Cohort (NFBC) dataset. NFBC enrolled almost everyone born in 1966 in Finland’s two most northern provinces. The NFBC dataset consists of 10 phenotypes and genotypes at 331,476 genetic variants measured in 5,327 individuals. We focus our study on the LDL cholesterol and triglyceride levels phenotypes.
Our evaluation strategy analyzes a subset of the NFBC data and compares what we discover here to what was discovered in the full NFBC dataset—which we treat as the gold standard. We compare the performance of our novel approach to three other methods: (1) the single univariate test applied to the disease status, (2) the multivariate approach applied to the disease status and the clinical phenotype modeled as a multivariate normal distribution, and (3) the liability threshold model treating the clinical phenotype as a covariate.
Using the univariate approach, the p-values are much weaker in comparison to those observed in the full NFBC dataset. Running the multivariate approaches, incorporating the triglyceride levels phenotypes, increased power (i.e., more significant p-values than SNPs).
Our method has the highest power in all scenarios. The advantage of our method is greater when there are substantial amounts of selection bias compared to lower amounts of selection bias. Our method is even more powerful when the correlation between the clinical covariate and the disease liability is lower, because we explicitly estimate the underlying liability using all of the data.
For more information, see our paper in Genetics: http://www.genetics.org/content/early/2017/01/27/genetics.116.198473
The software implementing the methods described in this paper was developed by Fernando Crespo and is available at: http://genetics.cs.ucla.edu/multipheno/ and
https://github.com/facrespo/BivariateProbitContinueEM
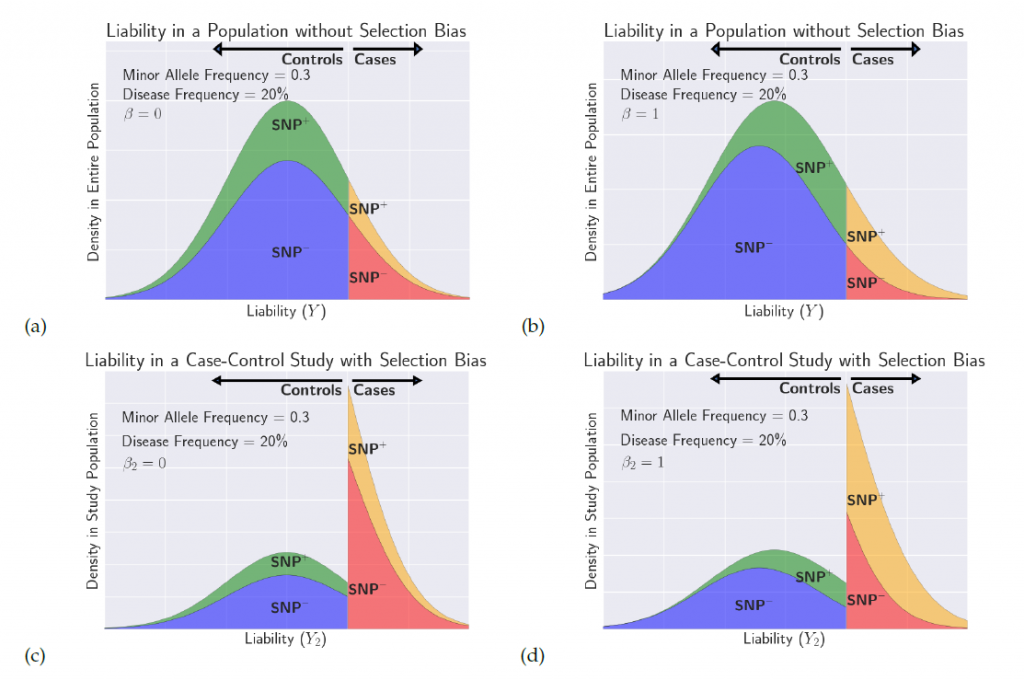
An illustration of the distribution of liability in a case-control study under selection bias. For more information, read our paper.
The full citation to our paper is:
Bilow, M., Crespo, F., Pan, Z., Eskin, E. and Eyheramendy, S., 2017. Simultaneous Modeling of Disease Status and Clinical Phenotypes to Increase Power in GWAS. Genetics, pp.genetics-116.