In a project led by Serghei Mangul, members of our lab recently developed and tested a novel computational method that uses regular RNA-Seq data to rapidly and accurately profile the human immune system. Mangul and his collaborators, including UCLA graduate student Harry (Taegyun) Yang and 2016 B. I. G. Summer undergraduate participants Jeremy Rotman, Benjamin Statz, and Will Van Der Wey, recently published their results in a paper on bioRxiv.
Discoveries in human immunology and advancements in development of treatments for many common human diseases depend on detailed reconstructions of the adaptive immune repertoire. The “adaptive” immune repertoire recognizes pathogens and toxins that the “innate” defense system misses. Assay-based genetic studies provide a detailed view of these adaptive systems by profiling the genetic expression and repertoires of B and T cell receptors. Assay-based approaches have accurately characterized the immune repertoire of peripheral blood.
However, these methods are expensive and smaller in scale when compared to standard RNA sequencing (RNA-seq). Characterizing the immunological repertoires of other tissues, including barrier tissues like skin and mucosae, requires large-scale study. RNA-Seq can capture the entire cellular population of a sample, including B and T cell and their receptors.
ImReP is the first method to efficiently extract B and T cell receptor derived reads from RNA-Seq data, accurately assemble CDR3 sequences, the most variable regions of these receptors, and determine their antigen specificity. Mangul and his team used simulated data to test the feasibility of using RNA-Seq to study the adaptive immune repertoire. ImReP is able to identify 99% CDR3-derived reads from the RNA-Seq mixture, suggesting it is a powerful tool for profiling RNA-Seq samples of immune-related tissues.
They also compared methods and investigated the sequencing depth and read length required to reliably assemble B and T cell receptor sequences from RNA-Seq data. ImReP consistently outperformed existing methods in both recall and precision rates for the majority of simulated parameters. Notably, ImReP was the only method with acceptable performance at 50bp read length, reconstructing with higher precision rate significantly more CDR3 clonotypes.
Mangul and his team applied ImReP to 8,555 samples across 544 individuals from 53 tissues obtained from Genotype-Tissue Expression study (GTEx v6). The data was derived from 38 solid organ tissues, 11 brain subregions, whole blood, and three cell lines. ImRep identified over 26 million reads overlapping 3.8 million distinct CDR3 sequences that originate from diverse human tissues.
Using ImReP, they created a systematic atlas of immunological sequences for B and T cell repertoires across a broad range of tissue types, most of which were not previously studied for B and T cell repertoires. They also examined the compositional similarities of clonal populations between tissues to track the flow of B and T clonotypes across immune-related tissues, including secondary lymphoid and organs encompassing mucosal, exocrine, and endocrine sites.
Advantages of using RNA-Seq to study immune repertoires include the ability to simultaneously capture both B and T cell clonotype populations during a single run, simultaneously detect overall transcriptional responses of the adaptive immune system, and scaling up the atlas of B and T cell receptors that will provide valuable insights into immune responses across various autoimmune diseases, allergies, and cancers.
Read more about ImReP in the full article, which is available for download on bioRxiv: http://biorxiv.org/content/early/2016/11/22/089235.article-metrics
ImReP was created by Igor Mandric and Serghei Mangul. ImReP is freely available at: https://sergheimangul.wordpress.com/imrep/
The atlas of T and B cell receptors, the largest collection of CDR3 sequences and tissue types, is freely available at https://sergheimangul.wordpress.com/atlas-immune-repertoires/. This resource has potential to enhance future studies in areas such as immunology and advance development of therapies for human diseases.
The full citation to our paper is:
Mangul, S., Mandric, I., Yang, H.T., Strauli, N., Montoya, D., Rotman, J., Van Der Wey, W., Ronas, J.R., Statz, B., Zelikovsky, A. and Spreafico, R., 2016. Profiling adaptive immune repertoires across multiple human tissues by RNA Sequencing. bioRxiv, p.089235.
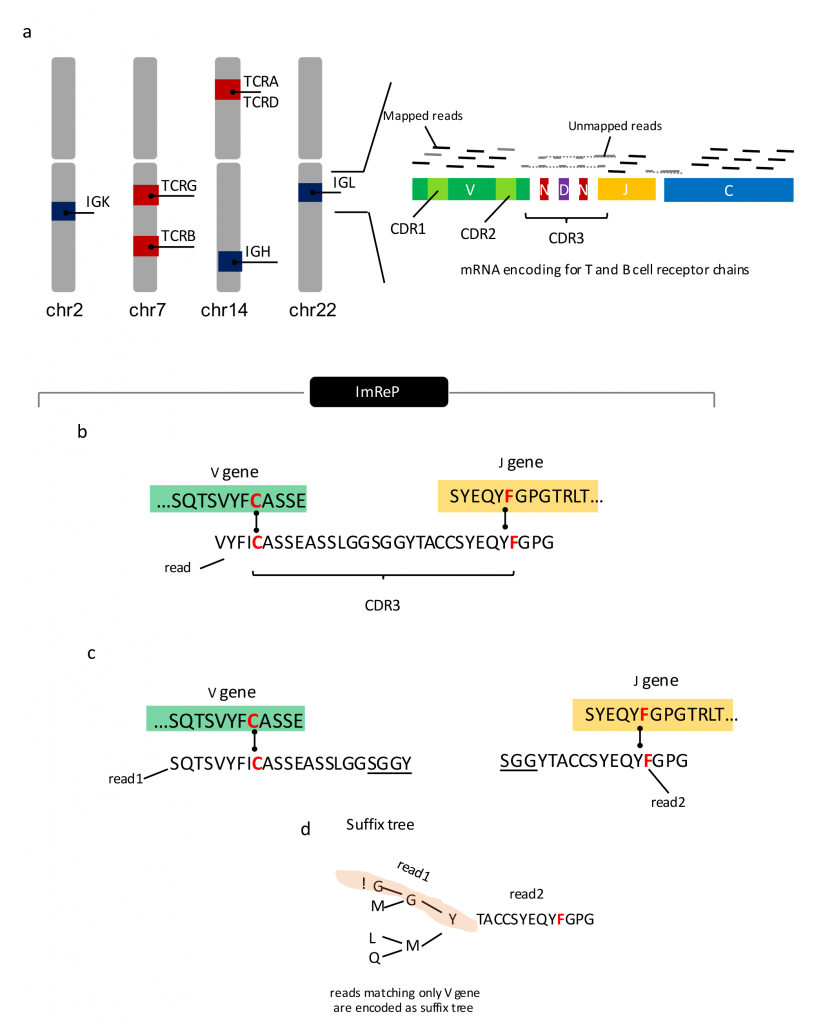
Figure 1. Overview of ImReP. (See full paper for details.)
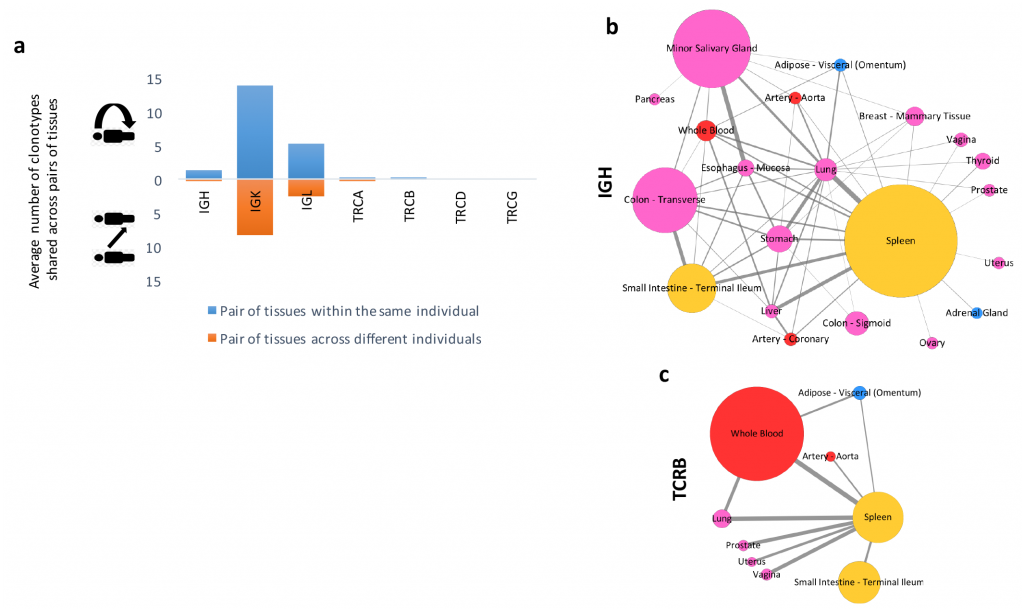
Figure 6. Flow of T and B cell clonotypes across diverse human tissues. (See full paper for details.)