Over the past several years, Genome Wide Association Studies (GWAS) have discovered hundreds of genetic variants involved in complex diseases(10.1056/NEJMra0905980). The vast majority of these variants do not lie in the protein coding regions of genes and thus do not affect what the gene produces, but instead likely affect how the genes are regulated. For this reason, the study of how genetic variation affect gene activity levels (referred to as expression levels) has been a major focus of research for many years. Genetic variation that affects gene expression are referred to as expression quantitative trait loci (eQTL)(10.1038/nrg2969).
Several studies collect expression from multiple tissues which leads to the question of whether or not the same genetic variants affect expression in multiple tissues(10.1038/ng.2653). Another way to ask this question is: Are eQTLs tissue specific or not tissue specific?
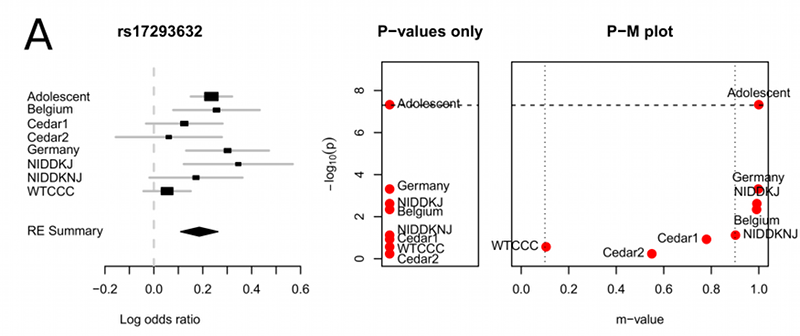
A challenge in this type of analysis is that an eQTL may affect expression in multiple tissues, but because of small sample sizes, the eQTL will only be detected in one of the tissues. Thus, traditional techniques for eQTLs will systematically be biased against detecting eQTLs in multiple tissues.
Jae-Hoon Sul and Buhm Han in our group developed a method to address this issue which builds upon recent methods in random effects meta-analysis(10.1016/j.ajhg.2011.04.014),(10.1371/journal.pgen.1002555). To apply these methods we first analyze each tissue separately and then use the meta-analysis method to combine the results of each tissue. Since our methods are specifically designed to handle “heterogeneity” which is that the effect size can be different in each study, our method is able to perform well when the effect is present in all of the tissues or just some of the tissues. More information about our meta-analysis research is here.
The full citation of our paper is here:
Over the past few years, our group has published several papers on methods for eQTL analysis. Our other paper on eQTL analysis include: