

Dat Duong, a graduate student in our lab, developed a novel method that will help find more eQTLs and eGenes in gene expression data from many tissues. A paper presenting his method is published in an upcoming issue of Bioinformatics.
Genome-wide association studies (GWAS) seek links between single-nucleotide polymorphisms (SNPs) and traits or diseases. SNPs are the most commonly occurring sources of variation in the human genome. Many SNPs identified by GWAS are located in intergenic regions, stretches of DNA sequences located between genes. SNPs identified in these primarily noncoding regions often do not have an obvious relationship to the disease phenotype. Other lines of evidence, such as gene expression, are required to explore this relationship and learn about disease function.
Gene expression, an intermediate phenotype between a causal SNP and a disease, can be used to interpret positive results produced by a GWAS. Common data types include expression quantitative trait loci (eQTLs), genetic variants associated with gene expression in particular tissue types, and eGenes, genes whose expression levels are associated with genetic variants. Both eQTL studies and GWAS focus on SNPs, but eQTL studies may provide biological insights into the disease development mechanism. For this reason, we pay special attention to the variants that are eQTLs or eGenes and have strong association signals identified by GWAS.
Multi-tissue gene expression datasets like the Gene Tissue Expression (GTEx) data are used to find eQTLs and eGenes. However, these datasets have small sample sizes in some tissues. Many meta-analysis methods have been designed to increase power for finding eQTLs and eGenes by combining gene expression data across many tissues However, these techniques cannot scale to datasets containing many tissue types, like the GTEx data. Such methods also ignore a biological principle that the same variant may be associated with the same gene across similar tissues.
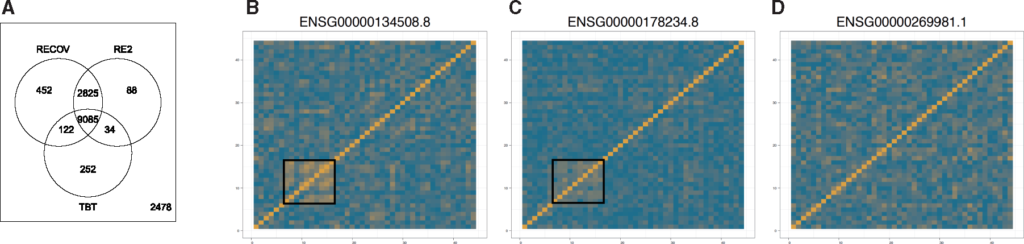
Venn diagram of the numbers of eGenes found by existing methods and RECOV, along with correlation matrices comparing methods. For more information, read our full paper.
To leverage the analytical power of eQTLs and eGenes in association studies, Duong and his team developed a new meta-analysis method named RECOV. Based on the principle that a SNP may have similar effect on the same gene in related tissues, RECOV can be applied to large gene expression datasets and can analyze all 44 tissues present in the GTEx data.
In our Bioinformatics paper, we use simulated datasets to show that RECOV has a correct false positive rate. When applied to real multi-tissue expression data from the GTEx dataset, RECOV detects 3% more eGenes than previous methods. RECOV is a general framework for meta-analysis that can be used with any COV matrix. We hope this software will be used by other researchers in the scientific community!
RECOV was developed by Dat Duong. The source code for RECOV is freely available at: https://github.com/datduong/RECOV.
Our paper can be downloaded at Bioinformatics: https://academic.oup.com/bioinformatics/article/33/14/i67/3953939/Applying-meta-analysis-to-genotype-tissue
The full reference for our paper is:
Duong, D., Gai, L., Snir, S., Kang, E.Y., Han, B., Sul, J.H. and Eskin, E., 2017. Applying meta-analysis to Genotype-Tissue Expression data from multiple tissues to identify eQTLs and increase the number of eGenes. Bioinformatics, 33(14), pp.i67-i74.
