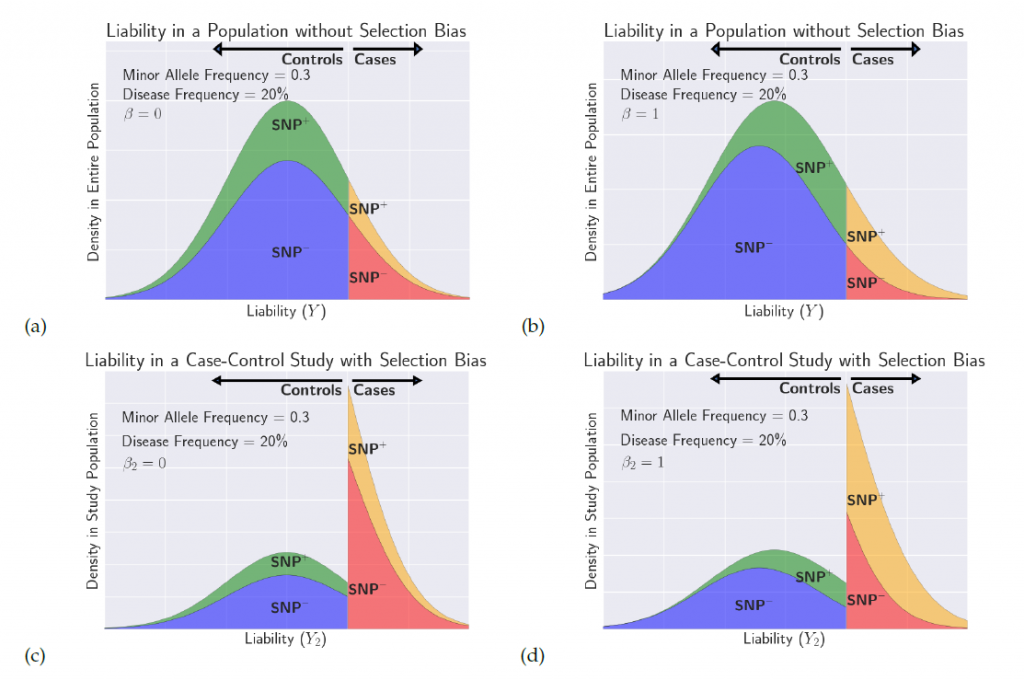
Variants regulating gene expression (expression quantitative trait loci, eQTL) are at a high frequency among SNPs associated with complex traits. Genome-wide characterization of gene expression is an important tool in genetic mapping studies of complex disorders, including many psychiatric disorders. Further, implicating eQTL to specific tissue types is key to understanding functional variation in disease development. Our group, in collaboration with Chiara Sabatti (Statistics, Stanford) and Nelson B. Freimer (David Geffen School of Medicine, UCLA), developed a novel approach for analyzing eQTL and applied the method to a dataset from a bipolar disorder study.
Current approaches to implicating eQTL specific to tissues lack sufficient power in large-scale studies of human brain related traits, such as bipolar disorder. Together with the University of California San Francisco, Universidad de Costa Rica, Universidad de Antioquia, Medellín, Colombia, and Tel Aviv University, our group adopted a novel approach to assess the heritability and genetic regulation of gene expression related to bipolar disorder in populations from Costa Rica and Colombia.
This project examines 786 genotyped subjects originally recruited in a study of bipolar disorder, all related within 26 extended families. While the subjects in this study were originally recruited as part of an investigation for severe bipolar disorder (BP1), we found no relationship between the observed gene expression data and BP1. Instead, we use this unique Latin American population to explore the architecture of genetic regulation. Specifically, we estimate heritability, evaluate the relative importance of local vs. distal genomic variation, identify variants with regulatory effects, and analyze the role of multiple associated SNPs in the same region.
Our group adopted a novel hierarchical testing procedure that leads to the analysis of eQTL data in a stage-wise manner with increasing levels of detail. This design allows us to compare estimates of the heritability of gene expression obtained using both traditional and genotype-based methods. First, we apply a multiscale testing strategy to identify SNPs that have regulatory effects (eSNPs) on BP1. Second, we investigate which specific probes are influenced by these eSNPs. This hierarchical testing procedure effectively controls error rates and leverages the heterogeneity across genetic variants to preserve computational power.
We use this approach to measure gene expression in lymphoblastoid cell lines (LCLs) in subjects from extended families, segregating for BP1. Our results suggest that variation in expression values is heritable and that, at least in samples including related individuals, relying on theoretical kinship coefficients or on realized genotype correlation for estimation of heritability leads to similar results.

Expression heritability and proportion of genetic variance due to local effects. For more information, see our paper. For more information, see our paper.
Variance decomposition approaches suggest that on average 30% of the genetic variance is due to local regulation. In the majority of probes under local regulation in our sample, more than one typed SNP is required to account for expression variation. This finding can be interpreted as the result of heterogeneity, but also could reflect un-typed causal variants that are tracked by more than one typed SNP.
The knowledge we acquired by studying the genetic regulatory network within these pedigrees, instead, can be used to inform our mapping studies: eSNPs might receive a higher prior probability of association, or be assigned a larger portion of the allowed global error rate when using a weighted approach to testing. We will report elsewhere on the results of these investigations.
For more information, see our paper, which is available for download through PLoS Genetics: http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1006046.
The full citation to our paper is:
Peterson, C.B., Jasinska, A.J., Gao, F., Zelaya, I., Teshiba, T.M., Bearden, C.E., Cantor, R.M., Reus, V.I., Macaya, G., López-Jaramillo, C. and Bogomolov, M., 2016. Characterization of Expression Quantitative Trait Loci in Pedigrees from Colombia and Costa Rica Ascertained for Bipolar Disorder. PLoS Genet, 12(5), p.e1006046.