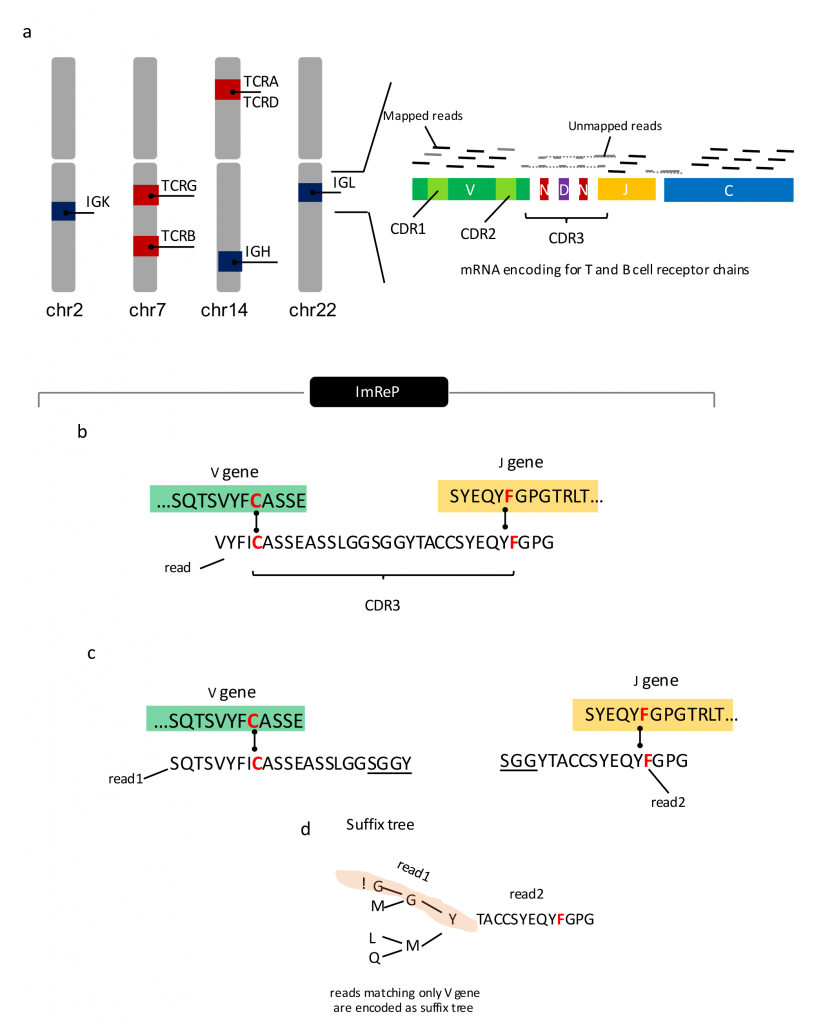
RNA viruses represent the majority of emerging and re-emerging diseases that pose a significant risk to global health – including influenza, hantaviruses, Ebola virus, and Nipah virus. When compared to DNA viruses, RNA viruses have an especially robust adaptability and evolvability due to their high mutation rates and rapid replication cycles. Development of novel medications for the prevention and treatment of these diseases requires an understanding of the mutant variants that drive an RNA-virus’ resistance mechanisms. The long read length offered by single-molecule sequencing technologies allows each mutant variant to be sequenced in a single pass. However, complete profiling of all viral genomes within a mutant spectrum is not yet possible due to the high error rate embedded in analytical protocols.
In collaboration with Alexander Artyomenko (Georgia State University), Alex Zelikovsky (Georgia State University), Nicholas Wu (The Scripps Research Institute), and Ren Sun (UCLA), Serghei Mangul and Eleazar Eskin developed a novel method for accurately reconstructing viral variants from single-molecule reads. This approach, two Single Nucleotide Variants (2SNV), tolerates the high error rate of the single molecule protocol and uses linkage between single nucleotide variations to efficiently distinguish these mutant variations from read errors.
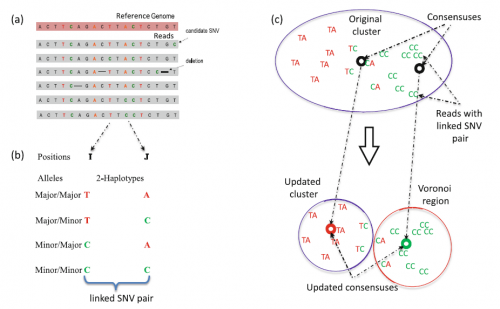
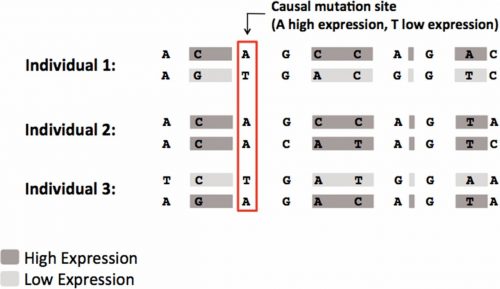
Overview of the 2SNV method. For more information, see our book chapter.
Any method for reconstructing viral variants from single-molecule reads must overcome low volume and high error rate of sequencing data, combined with very high similarity and very low frequency of viral variants. This challenge is similar to extraction of an extremely weak signal from very noisy background with signal-to-noise ratio approaching zero. However impossible this task may seem, a satisfactory solution can be based on distinguishing randomness of the noise from systematic signal repetition. With a high sensitivity and accuracy, 2SNV is anticipated to facilitate not only viral quasispecies reconstruction, but also other biological questions that require detection of rare haplotypes such as genetic diversity in cancer cell population, and monitoring B-cell and T-cell receptor repertoire.
We present 2SNV in a chapter of conference proceedings from the 2016 RECOMB meeting. To benchmark the sensitivity of 2SNV, we performed a single-molecule sequencing experiment on a sample containing a titrated level of known viral mutant variants. We tested 2SNV on a dataset comprised of PacBio reads from 10 independent clones, ranging from 1 to 13 mutations. These 10 clones were mixed at a geometric ratio with two-fold difference in occurrence frequency for consecutive clones starting with the maximum frequency of 50% and the minimum frequency of 0.1 %. Our method is able to accurately reconstruct clone with frequency of 0.2% and distinguish clones that differed in only two nucleotides distantly located on the genome. 2SNV outperforms existing methods for full-length viral mutant reconstruction.
For more information, see our book chapter, which is available for download through Springer Publications: http://link.springer.com/chapter/10.1007%2F978-3-319-31957-5_12.
In addition, the open source implementation of 2SNV, which was developed by Alexander Artyomenko, is freely available for download at http://alan.cs.gsu.edu/NGS/?q=content/2snv.
The full citation to our paper is:
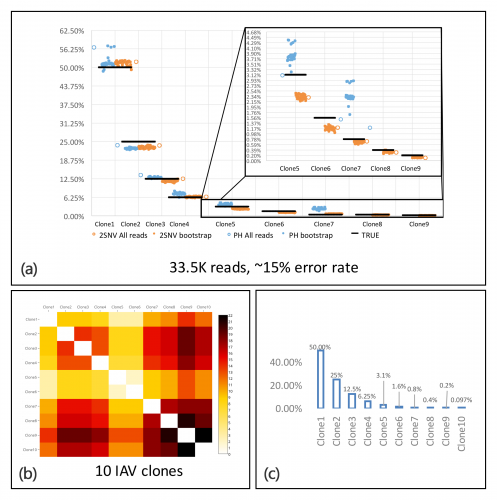
Overview of results using the 2SNV method. (a) 2SNV (orange) outperforms existing haplotype reconstruction tools (blue) in viral variant reconstruction. Using PacBio reads from 10 IAV clones, (b) the pairwise edit distance between clones given in a heat-map and (c) occurring frequency of clone types.